INTRODUCCIÓN
Las dermatosis neutrofílica son un grupo de trastornos caracterizados por lesiones en piel en las cuales el examen histológico revela infiltrados inflamatorios intensos epidérmicos y / o dérmicos compuestos principalmente por neutrófilos, sin evidencia de infección [1]. La clasificación de las dermatosis neutrofílica se basa en el reconocimiento de las características clínicas y patológicas, así como la identificación de enfermedades asociadas [1,2].
DERMATOSIS NEUTROFÍLICAS NO INFECCIOSAS Y SIN VASCULITIS
Compromiso Predominantemente Epidérmico:
- Psoriasis pustular.
- Pustulosis exantemática generalizada aguda inducida por drogas.
- Queratodermia blenorrágica.
- Dermatosis pustular subcórnea (enfermedad de Sneddon-Wilkinson).
- Pénfigo por IgA (dermatosis pustular tipo subcórnea, dermatosis neutrofílica intraepidérmica de tipo IgA)
- Pustulosis antimicrobiana de los pliegues.
- Acropustulosis infantil.
- Pustulosis neonatal transitoria.
Compromiso Predominantemente Dérmico:
- Síndrome de Sweet.
- Pioderma gangrenoso
- Enfermedad de Behçet.
- Síndrome artritis-dermatosis asociadas al intestino.
- Enfermedad inflamatoria intestinal (puede haber vasculitis de pequeños vasos)
- Hidradenitis écrinaneutrofílica .
- Dermatitis neutrofílica reumatoidea.
- Urticaria neutrofílica.
- Enfermedad de Still.
- Eritema marginado.
- Síndrome de fiebre periódica hereditaria.
Los hallazgos cutáneos en las dermatosis neutrofílicas son variables y pueden incluir vesículo-pústulas, placas, nódulos o úlceras [1,3,4]. Dependiendo del trastorno, las lesiones pueden ser localizadas o generalizadas. El compromiso extracutáneo puede estar presente en algunos casos.
La patogénesis de las dermatosis neutrofílica es desconocida. Se cree que estos trastornos representan un estado de reactividad inmunológica alterada. De acuerdo con esta hipótesis está la observación de que las dermatosis neutrofílica generalmente responden a los glucocorticoides sistémicos y otras terapias inmunomoduladoras [5].
SÍNDROME DE SWEET.
El prototipo de las dermatosis neutrofílica es el síndrome de Sweet (dermatosis neutrofílica aguda febril).El síndrome de Sweet tiene cuatro características fundamentales:
- Erupción cutánea que consiste en pápulas eritematosas y placas
- Infiltración dérmica neutrofílica no vasculítica en la biopsia
- Fiebre
- Neutrofilia en el frotis de sangre periférica [6,7].
La erupción cutánea consiste en pápulas eritematoso- violáceas dolorosas o sensibles a la palpación que se agrandan para formar placas con superficies irregulares pseudovesiculares. (Figuras 1 y 2)
![]()

Figura 1. Síndrome de Sweet.
Placa eritematosa con component pustular.

Figura 2. Síndrome de Sweet.
Placa brillante eritematosa con componente pustular presente en la cara de este paciente con síndrome de Sweet.
Pustulación verdadera y ampollas también pueden ocurrir. Las placas son por lo general de unos pocos centímetros de diámetro, y puede tener una coloración amarillenta central, creando una apariencia blanco-like. Las placas pueden causar dolor y sensación de quemazón, pero no son pruriginosas.
Aunque las lesiones de la piel puede ocurrir en cualquier sitio, se encuentran con mayor frecuencia en las extremidades, en la cara, el cuello y la parte superior, sobre todo en el dorso de las manos. Una variedad de manifestaciones sistémicas también puede ocurrir [6-9].
El síndrome de Sweet se asocia con una enfermedad subyacente en muchos pacientes [6]. Ejemplos de enfermedades y condiciones de base en síndrome de Sweet son: cáncer o enfermedades malignas en general, infecciones, drogas, enfermedades autoinmunes, enfermedad inflamatoria intestinal (Crohn, CU), y embarazo (Figuras 3,4,5,6).
![]()

Figura 3. Síndrome de Sweet.
Placas eritematosas en la cara de este paciente con síndrome de Sweet.

Figura 4. Síndrome de Sweet.
Placas con componente pustular en miembro superior.

Figura 5. Síndrome de Sweet
Placas inflamatorias presentes en extremidades.

Figura 6. Síndrome de Sweet.
Placas inflamatorias presentes en miembros superiores de este niño.
El síndrome de Sweet se clasifica en varias categorías:
- Síndrome de Sweet clásico
- Síndrome de Sweet asociado a neoplasias.
- Síndrome de Sweet inducido por medicamentos.
El síndrome de Sweet clásico o idiopático constituye la mayoría de los casos de síndrome de Sweet y se define como el síndrome de Sweet que cumple con los criterios de diagnóstico establecidos y no se asocia con malignidad ni a exposición a fármacos. Puede ocurrir en el contexto de una variedad de condiciones médicas como: infecciones de las vías respiratorias especialmente superiores e infecciones gastrointestinales, asociado a enfermedad inflamatoria intestinal (enfermedad de Crohn y colitis ulcerosa), y los asociados a embarazo. Asociaciones menos frecuentes son las relacionadas a la infección por el virus de la inmunodeficiencia humana [VIH], la tuberculosis, clamidias, hepatitis viral, inmunodeficiencias primarias, y enfermedades autoinmunes (por ejemplo, enfermedad de Behçet, policondritis recidivante, artritis reumatoide, sarcoidosis, enfermedades tiroideas autoinmunes, trastornos del tejido conectivo incluyendo lupus eritematoso sistémico y dermatomiositis). Son necesarios estudios adicionales para determinar la fuerza de las relaciones entre el síndrome de Sweet y estas enfermedades.
Síndrome de Sweet asociado a neoplasias.
Una revisión de 1993 de varias series retrospectivas encontró que 25 de 118 pacientes con síndrome de Sweet (21 por ciento) tenían una neoplasia hematológica o un tumor sólido asociados.
El síndrome de Sweet puede preceder, seguir o aparecer simultáneamente con una neoplasia. En los pacientes con antecedentes de cáncer, el desarrollo del síndrome de Sweet puede presagiar recurrencia de la enfermedad.
El síndrome de Sweet es más probable que ocurra en asociación con neoplasias malignas hematológicas, que con tumores sólidos. En una revisión de 1998 de 79 pacientes con neoplasias y el síndrome de Sweet, 69 (87 por ciento) había neoplasias malignas hematológicas y 12 tenían tumores sólidos (15 por ciento), incluyendo dos pacientes que tenían tanto la leucemia mielógena aguda (AML) y el cáncer de próstata.
Síndrome de Sweet inducido por medicamentos.
Varios medicamentos pueden contribuir al síndrome de Sweet : minociclina, nitrofurantoína, norfloxacina, ofloxacina, trimetoprima-sulfametoxazol, carbemazepina, diazepam, abacavir, bortezomib, imatinib, lenalidomida, clozapina, factores estimulantes de colonias, anticonceptivos, furosemida, diclofenac. Los más implicados en el desarrollo de síndrome de Sweet son los factores estimulantes de colonias granulocíticas. El síndrome de Sweet generalmente se desarrolla alrededor de dos semanas después de la exposición al fármaco en los pacientes que no tienen una historia previa de exposición a la droga incitar. La recurrencia del síndrome generalmente se desarrolla después de la reexposición al fármaco incitar.

Figura 7. Síndrome de Sweet.
Vesiculación en esta placa de un paciente con síndrome de Sweet.

Figura 8. Síndrome de Sweet.
Vesículas, pápulas y placas inflamatorias en este paciente con síndrome de Sweet.

Figura 9. Síndrome de Sweet.
Lesiones eritema-nodoso like en este paciente con síndrome de Sweet.
PIODERMA GANGRENOSO.
Pioderma gangrenoso (PG) es una enfermedad ulcerosa de la piel de origen desconocido. La primera lesión clínica es una pústula con una base inflamatoria, un nódulo eritematoso, o una ampolla hemorrágica con una base violácea (Figura 10).

Figura 10. Lesión temprana de pioderma gangrenoso.
Esta pústula con base eritematosa representa una lesión temprana de pioderma gangrenoso.
Estas lesiones evolucionan para formar úlceras superficiales o profundas, y pueden exponer a los tendones o los músculos subyacentes. Las úlceras tienen una base purulenta y un borde irregular, socavado, violáceo que se extiende periféricamente (figuras 11 y 12).

Figura 11. Pioderma gangrenoso.
Úlcera purulenta con borde irregular de color violáceo.

Figura 12. Pioderma gangrenoso.
Úlcera purulenta con borde irregular de color violáceo.
Si hay varias úlceras, estas pueden unirse para formar grandes lesiones que cuando curan lo hacen con atrofia, llamada cicatrización pigmentada cribiforme (Figura 13).

Figura 13. Pioderma gangrenoso.
Múltiples lesiones de pioderma gangrenoso en proceso de curación con cicatrización cribiforme pigmentada en una paciente con enfermedad inflamatoria intestinal.
PSORIASIS PUSTULOSA GENERALIZADA.
La psoriasis pustulosa generalizada es una dermatosis pustulosa aguda estéril, que se instala en pacientes con psoriasis en placas placa, eritrodermia psoriásica o psoriasis latente [10-13]. Se acompaña de signos y síntomas constitucionales, como fiebre, escalofríos, malestar general, mialgias y artralgias. El trastorno puede ocurrir a cualquier edad, incluso en niños [12,14,15]. La psoriasis pustulosa generalizada también puede ocurrir en el embarazo recibiendo el nombre de impétigo herpetiforme .
Hay varios factores precipitantes identificados [11-13]. Estos incluyen el embarazo, las infecciones, la hipocalcemia, salicilatos, yoduros, terapia con oro, litio, hidroxicloroquina, y la reducción o retirada de la terapia sistémica o local de glucocorticoides potentes. Hay dos casos en los que la psoriasis pustulosa generalizada siguió al uso de un ungüento que contiene calcipotriol y dipropionato de betametasona. [16].
Los factores genéticos también pueden estar involucrados en el desarrollo de este trastorno. Las mutaciones en el gen IL36RN han sido detectadas en algunos pacientes con psoriasis pustulosa generalizada [17,18].
La mayoría de los pacientes se presentan con grandes áreas de eritema doloroso que afecta a los sitios de flexión, regiones crural, y acral en el que múltiples pequeñas pústulas aparecen en grupos y se unen para formar lagos de pus (figuras 14, 14, 16, 17). Los problemas o complicaciones de esta entidad son la infección, la depleción de volumen, y un síndrome de fuga capilar posiblemente desencadenada por interleuquina-1 y factor de necrosis tumoral de los queratinocitos [19,20]. La afectación sistémica puede ocurrir, con manifestaciones tales como la artritis, e insuficiencias renal, hepática y cardiaca.

Figura 14. Psoriasis pustular.
Colección pustular en palmas de este paciente con psoriasis pustular.
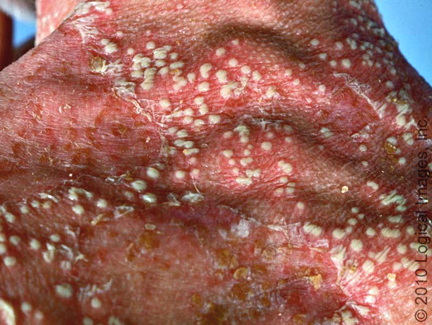
Figura 15. Pústulas en psoriasis pustular.

Figura 16. Psoriasis pustular.
Esta paciente de 28 años desarrolló fiebre, hipotensión, leucocitosis, eritema generalizado y pústulas a las 34 semanas de gestación durante su primer embarazo. Fue hospitalizada por varios meses y la erupción se resolvió en el primer mes después del parto de un niño normal a término. Una biopsia de piel mostró cambios típicos de psoriasis pustular.

Figura 17. Psoriasis pustular.
Numerosas pequeñas pústulas están presentes sobre placas eritematosas.
El diagnóstico diferencial de la psoriasis pustular generalizada incluye la pustulosis aguda eruptiva generalizada (PAEG) [21]. Como ambas pueden ser precipitada por las drogas (por ejemplo, betalactámicos y macrólidos) e infecciones por enterovirus, pueden ser confundidas. Las características clínicas de la pustulosis aguda eruptiva generalizada incluyen fiebre alta, erupción eritematosa y pustulosis (figuras 18 y 19). Otra manifestaciones cutáneas de la pustulosis aguda eruptiva generalizada pueden incluir eritema multiforme, vesículas, y púrpura. La histopatología muestra pústulas espongiformes subcorneales, vasculitis leucocitoclástica, e infiltración eosinofílica. El inicio es típicamente rápido y las lesiones cutáneas curan con la terapia de apoyo en dos a tres semanas.
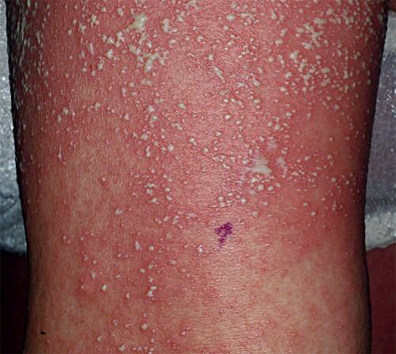
Figura 18. Pustulosis aguda eruptiva generalizada
Pústulas no foliculares confluentes sobre eritema edematoso en una mujer de 46 años con PAEG. Una biopsia de piel mostró pústulas con numerosos neutrófilos e infiltración neutrofílica de la epidermis y de la dermis superior.

Figura 19. Pustulosis aguda eruptiva generalizada
Pústulas no foliculares como una cabeza de alfiler con fondo edematoso y eritematoso son característicos de la PAEG.
El tratamiento de la GPP es en gran parte consiste en baños, emolientes y terapia sistémica. Entre los agentes sistémicos que se han utilizado con cierto éxito aparente son isotretinoína (40 a 80 mg/ día), la acitretina (25 mg / día), metotrexato (7,5 a 20 mg por semana), micofenolato mofetilo (1 gramo dos veces al día), y ciclosporina (3 a 5 mg / kg por día) [12,13]. Anti factor de necrosis tumoral alfa como infliximab y etanercept, se han reportado anecdóticamente eficaces en la psoriasis pustulosa generalizada [22,23]. Todavía se usa UVB de banda angosta o psoraleno oral más fototerapia UVA.
DEFICIENCIA DEL ANTAGONISTA DEL RECEPTOR DE IL-1.
Es un síndrome genético autoinflamatorio que se caracteriza por la aparición neonatal de osteomielitis multifocal estéril, periostitis, y pustulosis cutánea y que se ha descrito en varios pacientes [24-26]. La erupción cutánea se manifiesta por grupos de pústulas, pustulosis generalizada o lesiones ictiosiformes y puede ir acompañada de cambios en las uñas. El examen histopatológico de las lesiones cutáneas en pacientes afectados demuestra extensos infiltrados de neutrófilos tanto epidérmicos como dérmicos.
ARTRITIS REACTIVAS
La tríada de artritis, conjuntivitis y uretritis que ocurren después de una infección uretral o entérica es denominada artritis reactiva. Las lesiones genitales también son comunes. Veintitrés a cincuenta por ciento de los pacientes pueden tener balanitis. Balanitis aislada puede ser una forma frustra o puede proceder el desarrollo de otras características de la artritis reactiva [27].
La balanitis ocurre con mayor frecuencia en el margen coronal del prepucio y el glande, especialmente en el área perimeatal. La erupción se caracteriza por eritema que se desarrolla en forma de pápulas y pústulas erosivas. Las lesiones pueden coalescer y tienen bordes dentados, festoneados especialmente sobre el meato y forma la llamada "balanitis circinata" o balanitis circinada (Figura 21). Estas lesiones son indoloras en los pacientes no circuncidados y es necesario retraer el prepucio para detectar las lesiones. En los pacientes circuncidados, las lesiones pueden endurecerse y causar cicatrices o dolor. El diagnóstico diferencial de la balanitis debido a la artritis reactiva incluye eritema multiforme, erupción fija por drogas, psoriasis, herpes simple, candidiasis, y balanitis plasmocelular o enfermedad de Zoon.
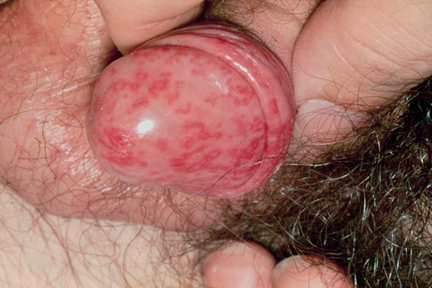
Figura 21.Balanitis circinada.
Erosiones eritematosas superficiales en distribución circinada presents en el glande de este paciente con artritis reactiva.
Una variedad de expresiones mucocutáneas de la enfermedad se pueden observar.
- Queratodermia blenorrágica, que se produce en el 10 al 30 por ciento de los casos, se encuentran en las plantas de los pies y las palmas de las manos, y son indistinguibles de la psoriasis pustular (Figura 22 y 23). Comienzan como vesículas claras sobre bases eritematosas y progresan a lesiones pustulosas queratósicas que confluyen para formar placas. Hay formación de absceso subungueal con la acumulación de restos y onicolisis (Figura 24).

Figura 22. Queratodermia blenorrágica.
Una visión cercana muestra placas descamativas de queratodermia blenorrágica en un paciente con artritis reactiva (antiguamente llamado síndrome de Reiter).

Figura 23. Queratodermia blenorrágica en artritis reactiva.
Múltiples vesículas y pústulas presentes en la planta del pie de este paciente con artritis reactiva.

Figura 24. Artritis reactiva.
Hiperqueratosis subungueal y onicolisis en artritis reactiva.
- Un estomatitis autolimitada caracterizada por ulceraciones superficiales o placas eritematosas grisáceas que involucran la mucosa bucal, paladar y lengua (Figura 25).

Figura 25. Lesiones orales en artritis reactiva.
Placas grisáceas presentes en la lengua
- Placas psoriasiformes puede aparecer en el cuerpo.
El tratamiento de las manifestaciones cutáneas con glucocorticoides tópicos, queratolíticos, o agentes sistémicos (metotrexato (2,5 a 20 mg por semana), micofenolato mofetil (1 gramo dos veces al día), o acitretina (25 mg / día) con o sin psoraleno más UVA (REPUVA ) pueden ser útiles. Mejoría en las manifestaciones de la piel y de las uñas se observó en un paciente con una enfermedad relacionada con el VIH que recibieó infliximab [28]. Otras terapias con anti- factor de necrosis tumoral alfa pueden ser eficaces y son: etanercept (25 mg SC dos veces por semana), adalimumab (40 mg s/c dos veces por semana) y talidomida (100 mg / día).
SÍNDROME SAPHO.
El reconocimiento de pacientes con características clínicas que incluyen: sinovitis, acné, pustulosis, hiperostosis y osteomielitis, llevó a Chamot a acuñar el acrónimo SAPHO [29]. El síndrome de SAPHO consta de un amplio espectro de dermatosis neutrofílicas asépticas asociadas a lesiones osteoarticulares también asépticas [30-32].
El trastorno generalmente afecta a niños y adultos jóvenes o de mediana edad. Los trastornos cutáneos asociados al síndrome SAPHO incluyen pustulosis palmar y plantar (Figura 26 ), folicular oclusión, acné fulminante, psoriasis pustular, enfermedad de Behçet, síndrome de Sweet, enfermedad de Sneddon-Wilkinson, pioderma gangrenoso, linear IgA bullous dermatosis bullosa por IgA, y la enfermedad de Lyme [33] .
http://www.elrincondelamedicinainterna.com/2008/12/ateneo-hospital-pintos-03122008.html
El trastorno generalmente afecta a niños y adultos jóvenes o de mediana edad. Los trastornos cutáneos asociados al síndrome SAPHO incluyen pustulosis palmar y plantar (Figura 26 ), folicular oclusión, acné fulminante, psoriasis pustular, enfermedad de Behçet, síndrome de Sweet, enfermedad de Sneddon-Wilkinson, pioderma gangrenoso, linear IgA bullous dermatosis bullosa por IgA, y la enfermedad de Lyme [33] .

Figura 26. Síndrome SAPHO.
Una mujer de 37 años con antecedentes de pustulosis palmo-plantar consultó por severo dolor torácico. La paciente fue primero diagnosticada de pustulosis palmo-plantar a los 36 años y rápidamente después del diagnóstico apareció el dolor torácico. Aunque el dolor y las lesiones de piel remitieron durante tres años después de escisión de la primera articulación esternocostal izquierda, ella experimentó recidiva del dolor esternal y de la pustulosis palmo-plantar. La medicación con prednisona, bucilamina, metotrexato y antibióticos fracasaron en el control de los síntomas. A los 45 años ella se sometió a amigdalectomía y sus síntomas se resolvieron y está asintomática hasta la actualidad sin pustulosis palmo-plantar ni dolor torácico.
PUSTULOSIS PALMOPLANTAR.
La pustulosis palmo-plantar se presenta como una vesiculopustulosis estéril con descamación, y eritema de las palmas de las manos y las plantas de los pies. A medida que las lesiones se van “secando”, se forman placas descamativas marrones hemorrágicas (Figura 27 y 28). La erupción suele ser crónica y simétrica y afecta principalmente a mujeres entre los 40 y 60 años de edad. Existen remisiones espontáneas y la enfermedad desaparece espontáneamente con el tiempo. Una posible asociación entre este trastorno y el uso de la anti-factor de necrosis tumoral-alfa, el anticuerpo monoclonal infliximab,para el tratamiento de la espondilitis anquilosante se ha visto [34].

Figura 27. Pustulosis palmo-plantar.
Pustulas con placas eritematosas descamativas.
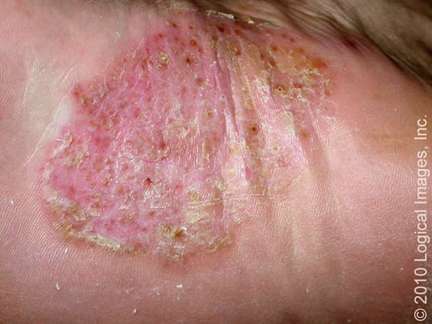
Figura 28. Pustulosis palmo-plantar.
Múltiples máculas marrones y pústulas presentes en una placa hiperqueratósica de la planta del pie.
La ubcación nosológica de la pustulosis palmo-plantar es incierta ya que se asocia a psoriasis en 15 a 30% de los casos y raramente a SAPHO. [35-37].
El tratamiento consiste en compresas, glucocorticoides tópicos, preparaciones de alquitrán, y terapias sistémicas. Los agentes sistémicos incluyen retinoides, tales como isotretinoína (40 a 80 mg / día) o acitretina (25 mg / día), metotrexato (2,5 a 20 mg por semana), biológicos agentes anti-TNF (adalimumab e infliximab), y PUVA ( Oxsoralen 10 a 40 mg / día) [38-43].
DERMATOSIS PUSTULAR SUBCÓRNEA O ENFERMEDAD DE SNEDDON WILKINSON.
La dermatosis pustular subcórnea o enfermedad de Sneddon-Wilkinson enfermedad fue descrita por primera vez en 1956 [44]. Este raro trastorno se ha observado en pacientes con artritis reumatoide, enfermedad inflamatoria intestinal, y tumores sólidos. Afecta prefernetemente a mujeres de entre 40 y 60 años.
La erupción se caracteriza por pústulas flácidas agrupadas en un patrón circinado sobre piel normal o eritematosa y se ve sobre todo en los pliegues inguinales y axilares, en el tronco, y en las partes proximales de las extremidades (Figuras 29 y 30). La histopatología revela una acumulación de neutrófilos subcórnea en ausencia de acantolisis (Figura 31).

Figura 29. Dermatosis pustular subcórnea o enfermedad de Sneddon-Wilkinson
Dermatosis pustular anular que afecta predominantemente áreas de pliegues causando pigmentación post inflamatoria en un paciente con enfermedad de Sneddon-Wilkinson

Figura 30.Dermatosis pustular subcórnea o enfermedad de Sneddon-Wilkinson.
Placas eritematosas anulares con pústulas superficiales están prsentes en la axila.

Figura 31. Dermatosis pustular subcórnea o enfermedad de Sneddon-Wilkinson.
Se observa una pústula neutrofílica subcórnea.
Generalmente no se presentan signos o síntomas sistémicos y la erupción es asintomática. Una variedad de asociaciones se han descrito. Estas incluyen una gammapatía monoclonal por IgA, crioglobulinemia, fenómeno de Raynaud [45], pioderma gangrenoso seguido por dermatosis pustular subcórnea [46], supuración de los ganglios linfáticos microbiana y abscesos asépticos bazo [47].
Generalmente no deja cicatriz pero una hiperpigmentación post-inflamatoria transitoria puede ocurrir. La enfermedad es relativamente benigna, pero a menudo tiene un curso crónico con recaídas. El diagnóstico diferencial clínico es el impétigo, la psoriasis pustulosa, pénfigo foliáceo, la dermatosis ampollosa lineal por IgA, la pustulosis exantemática, el eritema migratorio necrolítico y la pustulosis amicrobiana de los pliegues.
El tratamiento es dapsona (50 a 200 mg / día). Las terapias alternativas incluyen la acitretina (25 mg / día) PUVA, y la fototerapia UVB de banda estrecha [44,48].
SÍNDROME ARTRITIS-DERMATOSIS ASOCIADA AL INTESTINO.
Durante los años 1960 y 1970, se comenzó a llevar a cabo la cirugía de bypass yeyuno-ileal para el tratamiento de la obesidad mórbida. Aproximadamente el 20 por ciento de estos pacientes desarrollaron un síndrome similar a la gripe con una erupción cutánea episódica y recurrente [49]. Un síndrome similar se ha descrito en pacientes con enfermedad inflamatoria intestinal, y asociada a la operación de gastrectomía tipo Billroth II para tratamiento de la enfermedad ulcerosa péptica, la derivación biliopancreática, o asas ciegas intestinales como consecuencia de cirugías.[50].
Estos pacientes han recibido el diagnóstico desíndrome artritis-dermatosis asociada al intestino. Los pacientes afectados presentan un tipo enfermedad similar a la enfermedad del suero caracterizado por fiebre, escalofríos, malestar general, artralgias, artritis y mialgia, que suele preceder a la erupción cutánea. La poliartritis no erosiva aséptica es asimétrica, afecta episódicamente articulaciones grandes y pequeñas, y puede estar asociada con tenosinovitis. La diarrea y la mala absorción son características adicionales.
Las lesiones cutáneas más características de la piel son máculas eritematosas de hasta 1 cm de diámetro, que se desarrollan como una pápulo-vesícule central o pústula (Figura 32). La erupción se ve más frecuentemente en las extremidades superiores y el tronco, pero puede ocurrir en cualquier parte excepto las palmas de las manos, plantas de los pies, la cara y los genitales. Puede haber lesiones que recuerdan al eritema nodoso y se ha descripto también una paniculitis licuefaciente.

Figura 32. Dermatosis pápulo-pustular diseminada hemorrágica.
Lesiones pápulo-pustulares dispersas debidas a infiltración neutrofílica de la piel en un paciente con síndrome artritis-dermatosis por enfermedad inflamatoria intestinal.
La patogénesis es incierta. Sobrecrecimiento bacteriano intestinal con posterior formación de complejos inmunes en la circulación puede ser el evento inicial seguido por la deposición de estos complejos en un sitio del tejido blanco y la activación de la vía alterna del complemento.
Las opciones de tratamiento son reanastomosis en pacientes con cirugía de bypass, prednisona (10 a 60 mg / día) dapsona (100 mg / día), y antibióticos. Los antibióticos que pueden ser eficaces incluyen la tetraciclina (1-2 g / día), la minociclina (100 a 400 mg / día), clindamicina (600 mg / día), trimetoprim-sulfametoxazol (160/800 mg / día), eritromicina (500 a 2000 mg / día) y metronidazol (500 a 1000 mg / día) [49,51,52].
ENFERMEDAD DE BEHÇET
La enfermedad de Behçet es una enfermedad persistente y recurrente multisistémica con hallazgos mucocutáneos prominentes [53]. Se caracteriza por úlceras orales recurrentes en prácticamente todos los pacientes, asociado en muchos pacientes a lesiones genitales recurrentes, lesiones oculares, lesiones cutáneas y patergia.[54].
Las manifestaciones cutáneas se han dividido en dos categorías: un grupo inespecífico donde se ven foliculitis, acné, y dermografismo, y el llamado grupo neutrofílico –estéril donde se ven lesiones vesículo-pustulares, pústulas, lesiones pustulares hemorrágicas (vasculitis), placas y nódulos eritema nodoso-like (Figura 33) [55]. Las pápulas y placas eritematosas son edematosas a lesiones violáceas similares a las observadas en el síndrome de Sweet. Las pápulas purpúricas pueden llegar a ser pustulares.

Figura 33. Aftas orales en la enfermedad de Behçet.
Múltiples aftas dolorosas en un paciente con enfermedad de Behçet.
Las lesiones nodulares eritematosas que se asemejan al eritema nudoso, involucran piernas, glúteos, brazos y cuello. Estas lesiones difieren del eritema nodoso por su duración más corta (de una a tres semanas), menos induración, y una distribución diferente, así como la imagen histopatológica. Sin embargo, eritema nodoso y vasculitis cutánea también se han visto en la enfermedad de Behçet (
Patergia se refiere a una pápula eritematosa o pustular respuesta a una lesión cutánea local. Se define como una lesión mayor que 5 mm que aparece 24 a 48 horas después de la punción de la piel con una aguja.
El hallazgo característico histopatológico de las lesiones tempranas es un infiltrado neutrofílico angiocéntrico con vasculitis leucocitoclástica. Las lesiones tardías muestran una infiltración linfocítica.
Las manifestaciones mucocutáneas de la enfermedad de Behçet puede ser tratada con colchicina oral (0,6 mg administrada dos a tres veces al día), anestésicos tópicos o intralesionales, o glucocorticoides sistémicos. La talidomida (200 mg / día) y dapsona (100 a 200 mg / día) también han demostrado eficacia [56]. Otros fármacos que se han reportado eficaces son interferón-2a, azatioprina, ciclosporina, ciclofosfamida, inmunoglobulina intravenosa, e inhibidores del factor de necrosis tumoral-alfa (etanercept e infliximab).
HIDRADENITIS ÉCRINA NEUTROFÍLICA.
La hidradenitis écrina neutrofílica es un trastorno reactivo que puede ocurrir en asociación con cáncer (con y sin quimioterapia), infecciones y/o ciertos medicamentos [57,58]. Las lesiones clínicas son típicamente placas eritematosas edematosas que pueden ser purpúrica y dolorosas. Están situadas en las extremidades, el tronco, la cara y las palmas, y pueden imitar celulitis cuando están presentes cerca de los ojos. Fiebre y neutropenia pueden ser características concurrentes.
La histopatología revela infiltrado neutrofílico que rodea la glándula écrina con formación ocasional abscesos intraductales y necrosis de las células secretoras.
Los tumores malignos asociados incluyen leucemia mieloide, otras leucemias, linfomas de Hodgkin y tumores sólidos. El compromiso de las glándulas sudoríparas puede ocurrir en ausencia de exposición a quimioterapia (bleomicina, clorambucilo, ciclofosfamida, cytarabina, doxorrubicina, lomustima, mitoxantrona, topotecan, y vincristina). Entre los agentes infecciosos reportados en asociación con este trastorno de la piel están Serratia marcescens, Enterobacter cloacae, Staphylococcus aureus y el virus de la inmunodeficiencia humana. Además de los agentes quimioterápicos, otros fármacos posiblemente asociados son paracetamol, zidovudina, estavudina, factor estimulante de colonias granulocíticas, y minociclina.
Las lesiones generalmente se resuelven espontáneamente dentro del mes, pero pueden reaparecer. La dapsona se ha utilizado y puede ser útil [59].
DERMATOSIS NEUTROFÍLICA DEL DORSO DE LAS MANOS.
La dermatosis neutrofílica del dorso de las manos (vasculitis pustular) es considerada por algunos autores como una variante del síndrome de Sweet, pero la presencia de vasculitis leucocitoclástica en algunos casos, por lo demás similares ha llevado a que otros la clasificaran como una " vasculitis pustular"[60 ,61].
Las características clínicas son nódulos y placas dolorosos situados en la cara dorsal de las manos, los cuales pueden pasar a formar pústulas y /o úlceras (Figura 34) [60,62-64]. Aunque la presentación inicial es sugestiva de infección cutánea, las lesiones no responden al tratamiento con antibióticos y los cultivos microbiológicos y examen histológico con manchas de bacterias, micobacterias y hongos han sido negativos. Las mujeres son más afectadas que los hombres.

Figura 34. Dermatosis neutrofílica del dorso de las manos.
Placas dolorosas que progresan a la ulceración que aparecieron en una mujer de 60 años con artritis reumatoidea seropositiva. La apariencia histológica fue la hiperplasia epitelial con abscesos estériles intraepiteliales y denso infiltrado dérmico y ausencia de vasculitis leucocitoclástica.
El informe inicial de esta entidad observó vasculitis leucocitoclástica en la piel afectada [60], sino una descripción posterior de los pacientes con similares hallazgos físicos sólo había infiltrado dérmico constituido por neutrófilos y pústulas en el examen microscópico [62]. En una serie de siete pacientes con este trastorno, sólo uno tenía evidencia de vasculitis [64].
DERMATOSIS NEUTROFÍLICA ASOCIADA A ARTRITIS REUMATOIDEA.
Síndrome de Sweet, pioderma gangrenoso, dermatitis neutrofílica reumatoide y dermatitis granulomatosa neutrofílica en empalizada pueden aparecer en pacientes con artritis reumatoide. Otros tipos de lesiones de la piel que no están clasificados entre las dermatosis neutrofílicas también pueden estar asociados con la artritis reumatoide.
Dermatitis neutrofílica reumatoidea.
La dermatitis neutrofílica reumatoidea fue descripta por primera vez en 1978. Menos de 10 casos se han comunicado [65-68]. Se presenta con mayor frecuencia en mujeres de mediana edad con AR grave seropositivas. La erupción consiste típicamente en pápulas, placas o nódulos eritematosas uticarianas, y menores de 2 cm de diámetro [66,67]. Las lesiones suelen ser simétricas y predominan en las extremidades, cuello y tronco, que no suelen ser pruriginosas pero sí dolorosas.
Histopatológicamente, hay un denso infiltrado neutrofílico dérmico sin vasculitis. Puede haber leucocitoclasia, y el infiltrado puede extenderse al tejido subcutáneo. La papila dérmica puede tener microabscesos [69].
A diferencia de síndrome de Sweet, las lesiones suelen ser asintomáticas, tienden a resolverse espontáneamente o con la mejoría de la artritis reumatoide después de una a tres semanas, carecen de los signos y síntomas constitucionales como fiebre y malestar y enfermedad ocular [68]. La terapia consiste en glucocorticoides tópicos o sistémicos, dapsona (100 a 200 mg / día), y antimaláricos, como hidroxicloroquina (200 mg 2 x día) [68].
Dermatitis granulomatosa neutrofílica en empalizada.
La dermatitis granulomatosa en el contexto de la enfermedad autoinmune presenta en dos patrones principales clínicas e histopatológicas, dermatitis granulomatosa neutrofílica en empalizada (DGNE) (Figura 35) y la dermatitis granulomatosa intersticial (DGI)[70,71,72]. Otros nombres que se han utilizado para describir a la DGNE en la literatura incluyen pápulas reumatoides, granuloma de Churg-Strauss, granuloma cutáneo necrosante extravascular y necrobiosis ulcerosas reumatoidesuperficial.

Figura 35. Dermatitis granulomatosa neutrofílica en empalizada.
La DGNE se puede presentar como pápulas umbilicadas simétricamente distribuidas que favorecen las superficies extensoras de las extremidades, particularmente los codos y los dedos. La asociación con enfermedad más frecuentes con la artritis reumatoide, pero también pueden aparecer en el lupus eritematoso sistémico, en las vasculitis sistémicas vasculitis sistémica, y en otros trastornos autoinmunes [73]. Se plantea la hipótesis de que la deposición de complejos inmunes en los vasos sanguíneos puede ser el factor desencadenante[70]. Los hallazgos histopatológicos de las lesiones tempranas incluyen una vasculitis leucocitoclástica de pequeños vasos y un denso infiltrado neutrofílico [70]. Lesiones mayores presentan granulomas en empalizada y fibrosis dérmica.
El término dermatitis granulomatosa intersticial describe una forma de dermatitis granulomatosa en la que las placas anulares en el tronco de pacientes con artritis reumatoidea (Figura 36) [74,75].

Figura 36. Dermatitis granulomatosa intersticial.
Placas anulares eritematosas en el tronco de este paciente con AR.
La dermatitis granulomatosa intersticial ha estado asociada con una variedad de factores incluyendo las enfermedades autoinmunes, cáncer, y drogas [76]. Los hallazgos histopatológicos incluyen focos de colágeno degenerado con granulomas en empalizada compuesto por histiocitos. Esto se acompaña de in infiltrado dérmico intersticial linfohistiocitario con número variable de neutrófilos y eosinófilos. No hay vasculitis [33,72].
El diagnóstico clínico diferencial de la forma papular de DGNE incluye el granuloma anular papular, y los nódulos reumatoides [33].
El tratamiento es con corticosteroides tópicos de alta potencia, corticosteroides intralesionales, dapsona [78], e hidroxicloroquina [79]. La dermatitis granulomatosa intersticial y la DGNE han ocurrido después de la iniciación de la terapia anti-TNF [80,81]. Sin embargo, la dermatitis granulomatosa intersticial también se ha mejorado con fármacos anti-TNF [82,83].
REFERENCIAS
1.Callen JP. Dermatosis neutrofílica. Dermatol Clin 2002; 20:409.
2.James WD. Entrada más reciente dermatosis neutrofílica. Arch Dermatol 2003; 139:101.
3.Jorizzo JL Salomón, AR, Zanolli MD, Leshin B. reacciones vasculares neutrofílica. J Am Acad Dermatol 1988; 19:983.
4.Moschella SL. Examen de llamadas dermatosis neutrofílica asépticas. Australas J Dermatol 1983; 24:55.
5.Cohen PR. Dermatosis neutrofílicas: una revisión de las opciones de tratamiento actuales. Am J Clin Dermatol 2009; 10:301.
6.Kemmett D, Hunter JA. El síndrome de Sweet: una revisión clínico-patológicas de veintinueve casos. J Am Acad Dermatol 1990; 23:503.
Síndrome 7.von den Driesch P. Sweet (dermatosis neutrofílica febril aguda). J Am Acad Dermatol 1994; 31:535.
8.Moreland LW, Brick JE, Kovach RE, et al. Dermatosis neutrofílica febril aguda (síndrome de Sweet): una revisión de la literatura con énfasis en las manifestaciones musculoesqueléticas. Semin Arthritis Rheum 1988; 17:143.
9.Hisanaga K, Iwasaki Y, Itoyama Y, Neuro-Sweet Grupo de Estudio de Enfermedades. Neuro-Sweet enfermedad: manifestaciones clínicas y criterios para el diagnóstico. Neurología 2005; 64:1756.
10.Baker H, Ryan TJ. Psoriasis pustulosa generalizada. Un estudio clínico y epidemiológico de 104 casos. Br J Dermatol 1968; 80:771.
11.Baker H. Generalizado psoriasis pustulosa. En: Psoriasis, Roenigk HH Jr, Maibach HI (Eds), Marcel Dekker, 1985. p.15.
12.Zelickson BD, Muller SA. Psoriasis pustulosa generalizada. Una revisión de 63 casos. Arch Dermatol 1991; 127:1339.
13.Farber EM, Nall L. psoriasis pustulosa. Cutis 1993, 51:29.
14.de Oliveira ST, Maragno L, Arnone M, et al. Psoriasis pustulosa generalizada en la infancia. Pediatr Dermatol 2010; 27:349.
15.Zelickson BD, Muller SA. Psoriasis pustulosa generalizada en la infancia. Informe de trece casos. J Am Acad Dermatol 1991; 24:186.
16.Tobin AM, SM Langan, Collins P, B. Kirby psoriasis pustulosa generalizada (von Zumbusch) tras el uso de calcipotriol y dipropionato de betametasona pomada: a propósito de dos casos. Clin Exp Dermatol 2009; 34:629.
17.Onoufriadis A, Simpson MA, Rosa AE, et al. Las mutaciones en IL36RN/IL1F5 están asociados con la grave enfermedad episódica inflamatoria de la piel conocida como psoriasis pustular generalizada. Am J Hum Genet 2011; 89:432.
18.Marrakchi S, P Guigue, Renshaw BR, et al. La interleucina-36-antagonista del receptor de la deficiencia y la psoriasis pustulosa generalizada. N Engl J Med 2011; 365:620.
19.McGregor JM, Barker JN, MacDonald DM. El síndrome de fuga capilar pulmonar que complica la psoriasis pustulosa generalizada: el posible papel de las citocinas. Br J Dermatol 1991; 125:472.
20.Handfield-Jones SE, Garvey M, McGibbon DH, MM Negro. Síndrome de fuga capilar en la psoriasis pustulosa generalizada. Br J Dermatol 1992; 127:64.
21.Beylot C, Bioulac P, Doutre MS. [Agudas generalizadas pustuloses exantemáticas (cuatro casos) (transl autor)]. Ann Dermatol Venereol 1980; 107:37.
22.Williams JD, Griffiths CE. Los agentes bloqueadores de citoquinas en dermatología. Clin Exp Dermatol 2002; 27:585.
23.Kamarashev J, Lor P, Forster A, et al. Psoriasis pustulosa generalizada inducida por la ciclosporina una retirada en respuesta a la necrosis tumoral etanercept inhibidor de factor alfa. Dermatology 2002; 205:213.
24.Aksentijevich I, Masters SL, Ferguson PJ, et al. Una enfermedad autoinflamatoria con deficiencia del antagonista de interleucina-1-receptor. N Engl J Med 2009; 360:2426.
25.Reddy S, S Jia, Geoffrey R, et al. Una enfermedad autoinflamatoria debido a la deleción homocigota del locus IL-1RN. N Engl J Med 2009; 360:2438.
26.Minkis K, Aksentijevich I, Goldbach-Mansky R, et al. La interleucina 1 antagonista del receptor de la deficiencia se presenta como pustulosis infantil imitando psoriasis pustulosa infantil. Arch Dermatol 2012; 148:747.
27.Calin A. Taller III. Tratamiento del síndrome de Reiter. Ann Rheum Dis 1979; 38 Suppl 1: supl 96.
28.Gaylis N. Infliximab en el tratamiento de un paciente VIH positivo con el síndrome de Reiter. J Rheumatol 2003; 30:407.
29.Chamot AM, Benhamou CL, Kahn MF, et al. [Acné-pustulosis-hiperostosis, osteítis síndrome. Resultados de una encuesta nacional. 85 casos]. Rev Rhum Osteoartic 1987 Mal; 54:187.
30.Kahn MF, MA Khan. El síndrome de SAPHO. Baillieres Clin Rheumatol 1994; 8:333.
31.Beretta-Piccoli BC, Sauvain MJ, Gal I, et al. La sinovitis, acné, pustulosis, hiperostosis, osteítis (SAPHO) síndrome en la infancia: un informe de cada diez casos y revisión de la literatura. Eur J Pediatr 2000; 159:594.
32.Van Doornum S, Barraclough D, G McColl, Wicks I. SAPHO: raro o simplemente no reconocido? Semin Arthritis Rheum 2000; 30:70.
33.Rencic A, Nousari CH. Otras enfermedades reumatológicas. En: Dermatología, 2 ª ed, Bolognia JL, JL Jorizzo, RP Rapini, et al (eds), Elsevier Limited, España 2008. p.597.
34.Baeten D, E Kruithof, Van den Bosch F, et al. Seguridad sistemático de seguimiento de una cohorte de 107 pacientes con espondiloartropatía tratados con infliximab: una nueva perspectiva sobre el papel de la defensa del huésped en la patogenia de la enfermedad? Ann Rheum Dis 2003; 62:829.
35.Saghafi M, MJ Henderson, Buchanan WW. Hiperostosis Esternocostoclavicular. Semin Arthritis Rheum 1993; 22:215.
36.Kahn MF, Chamot AM. SAPHO síndrome. Rheum Dis Clin North Am 1992; 18:225.
37.Paller AS, Pachman L, Rich K, et al. Pustulosis palmar plantar et: su asociación con osteomielitis crónica multifocal recurrente. J Am Acad Dermatol 1985; 12:927.
38.Reymann F. Dos años de experiencia con el tratamiento Tigason de pustulosis palmo-plantar y manuum keratoticum eczema. Dermatologica 1982; 164:209.
39.Thomsen K. Pustulosis palmar plantar et tratados con metotrexato. Acta Derm Venereol 1971; 51:397.
40.Morison WL, Parrish JA, TB Fitzpatrick. Oral fotoquimioterapia methoxsalen de dermatosis recalcitrantes de las palmas y las plantas de los pies. Br J Dermatol 1978; 99:293.
41.Marsland AM, Chalmers RJ, Hollis S, et al. Intervenciones para la psoriasis pustulosa palmoplantar crónica. Cochrane Database Syst Rev. 2006;: CD001433.
42.Yawalkar N, RE Hambre. El éxito del tratamiento de la psoriasis recalcitrante palmoplantar pustulosa con el uso secuencial de infliximab y adalimumab. Dermatology 2009; 218:79.
Lckova-Laskoska 43.V 'MT, Caca-Biljanovska NG, Laskoski DS, Kamberova SJ. Pustulosa palmoplantar tratados con itraconazol: un único activo-brazo del estudio piloto. Dermatol Ther 2009; 22:85.
44.Sneddon IB, Wilkinson DS. Subcorneal Dermatosis pustulosa. Br J Dermatol 1979; 100:61.
45.Kasha EE Jr, Epinette WW. Subcorneal pustular dermatosis (Sneddon-Wilkinson enfermedad) en asociación con una gammapatía monoclonal IgA: un clínico y revisión de la literatura. J Am Acad Dermatol 1988; 19:854.
46.Kohl PK, Hartschuh W, W Tilgen, PJ Frosch. Pioderma gangrenoso seguido de dermatosis pustulosa subcorneal en un paciente con paraproteinemia IgA. J Am Acad Dermatol 1991; 24:325.
47.Dallot A, Decazes JM, Drouault Y, et al. Subcorneal pustular dermatosis (Sneddon-Wilkinson enfermedad) con supuración de los ganglios linfáticos y amicrobiana abscesos asépticos bazo. Br J Dermatol 1988; 119:803.
48.Todd DJ, Bingham EA, M Walsh, D. Burrows subcorneal pustular dermatosis IgA y paraproteinemia: La respuesta a ambas etretinato y PUVA. Br J Dermatol 1991; 125:387.
49.Kennedy C. El espectro de la enfermedad inflamatoria de la piel después de bypass yeyuno-ileal para obesidad mórbida. Br J Dermatol 1981; 105:425.
50.Dicken CH. Intestinal asociada a dermatosis-artritis síndrome: el síndrome del intestino de bypass sin bypass intestinal. J Am Acad Dermatol 1986; 14:792.
51.Stein HB, Schlappner OL, Boyko W, et al. El bypass intestinal: síndrome de artritis-dermatitis. Arthritis Rheum 1981; 24:684.
PH 52.Ely. El síndrome del intestino derivación: una respuesta a peptidoglicanos bacterianos. J Am Acad Dermatol 1980; 2:473.
53.Chajek T, enfermedad de Behçet Fainaru M.. Informe de 41 casos y revisión de la literatura. Medicine (Baltimore) 1975; 54:179.
54.Criteria para el diagnóstico de la enfermedad de Behçet. Grupo de Estudio Internacional para la Enfermedad de Behçet. Lancet 1990; 335:1078.
55.Arbesfeld SJ, Kurban AK. Enfermedad de Behçet. Nuevas perspectivas sobre un síndrome enigmático. J Am Acad Dermatol 1988; 19:767.
56.Hamuryudan V, Mat C, Saip S, et al. La talidomida en el tratamiento de las lesiones mucocutáneas del síndrome de Behçet. Un estudio aleatorizado, doble ciego, controlado con placebo. Ann Intern Med 1998; 128:443.
57.Harrist TJ, Fine JD, Berman RS, et al. Hidradenitis neutrofílica ecrino. Un tipo distinto de dermatosis neutrofílica asociado con la leucemia mielógena y quimioterapia. Arch Dermatol 1982; 118:263.
58.Roustan G, Salas C, Cabrera R, Simón A. hidradenitis neutrofílica ecrino no asociada con la quimioterapia en pacientes con leucemia mieloide aguda. Int J Dermatol 2001; 40:144.
59.Shear NH, SR Knowles, L Shapiro, Põldre P. dapsona en la prevención de la recurrencia ecrino hidradenitis neutrofílica. J Am Acad Dermatol 1996; 35:819.
60.Strutton G, D Weedon, Robertson I. vasculitis pustulosa de las manos. J Am Acad Dermatol 1995; 32:192.
61.Cohen PR. Las lesiones cutáneas del síndrome de Sweet y su variante mano dorsal contiene vasculitis: una contradicción o un epifenómeno? Arch Dermatol 2002; 138:400.
62.Galaria NA, JM Junkins-Hopkins, D Kligman, WD James. Dermatosis neutrofílica del dorso de las manos: pustular revisited vasculitis. J Am Acad Dermatol 2000; 43:870.
63.Curcó N, Pagerols X, Tarroch X, P. Vives vasculitis pustulosa de las manos. Presentación de dos hombres. Dermatology 1998; 196:346.
64.DiCaudo DJ, Connolly SM. Dermatosis neutrofílica (vasculitis pustular) de las manos dorsales: reporte de 7 casos y revisión de la literatura. Arch Dermatol 2002; 138:361.
65.Ackerman AB. El diagnóstico histopatológico de la enfermedad de la inflamación cutánea. Un método de análisis de patrones, Lea y Febiger, Philadelphia 1978. p.444.
66.Scherbenske JM, PM Benson, GP Lupton, CP Samlaska. Dermatitis neutrofílica reumatoide. Arch Dermatol 1989; 125:1105.
67.Mashek HA, Pham TC, TN Helm, Klaus M. dermatitis neutrofílica reumatoide. Arch Dermatol 1997; 133:757.
68.Sánchez JL, Cruz A. dermatitis neutrofílica reumatoide. J Am Acad Dermatol 1990; 22:922.
69.Lowe L, Kornfeld B, Clayman J, Golitz LE. Dermatitis neutrofílica reumatoide. J Cutan Pathol 1992; 19:48.
70.Chu P, Connolly MK, LeBoit PE. El espectro histopatológico de dermatitis granulomatosa palisaded neutrofílica y en pacientes con enfermedad vascular del colágeno. Arch Dermatol 1994; 130:1278.
71.Sangueza OP, Caudell MD, Mengesha YM, et al. Empalizada dermatitis granulomatosa neutrofílica en la artritis reumatoide. J Am Acad Dermatol 2002; 47:251.
72.Long D, DM Thiboutot, JT Majeski, et al. Dermatitis granulomatosa intersticial con artritis. J Am Acad Dermatol 1996; 34:957.
73.Gulati A, D Paige, Yaqoob M, et al. Dermatitis neutrofílica empalizada granulomatosa asociada a lupus eritematoso sistémico presentan con el signo de la cuerda en llamas. J Am Acad Dermatol 2009; 61:711.
74.Tomasini C, Pippione dermatitis granulomatosa intersticial con M. placas. J Am Acad Dermatol 2002; 46:892.
75.Verneuil L, Dompmartin A, Comoz F, et al. Dermatitis granulomatosa intersticial con cuerdas cutáneas y artritis: un trastorno asociado con autoanticuerpos. J Am Acad Dermatol 2001; 45:286.
76.Heidary N, S Mengden, Pomeranz MK. Empalizada dermatosis neutrofílica y granulomatosa. J Dermatol Online 2008; 14:17.
77.Germanas JP, D Mehrabi, KR Carder. Empalizada dermatitis granulomatosa neutrofílica en una niña de 12 años con lupus eritematoso sistémico. J Am Acad Dermatol 2006; 55: S60.
78.Martin JA, Jarrett pápulas P. reumatoide tratados con dapsona. Clin Exp Dermatol 2004; 29:387.
79.Gerbing EK, D Metze, Luger TA, Ständer S. [dermatitis granulomatosa intersticial sin artritis: La terapia exitosa con hidroxicloroquina]. Dtsch Ges J Dermatol 2003; 1:137.
80.Deng A, Harvey V, Sina B, et al. Dermatitis granulomatosa intersticial asociada con el uso de inhibidores del factor de necrosis tumoral alfa. Arch Dermatol 2006; 142:198.
81.Bremner R, Simpson E, White CR, et al. Empalizada dermatitis neutrofílica y granulomatosa: una manifestación cutánea poco común de los trastornos autoinmunes. Semin Arthritis Rheum 2004; 34:610.
82.Zoli A, G Massi, Pinnelli M, et al. Dermatitis granulomatosa intersticial en la artritis reumatoide responder a etanercept. Clin Rheumatol 2010; 29:99.
83.Kreuter A, Gambichler T, Altmeyer tratamiento con infliximab P. de la dermatitis granulomatosa intersticial. J Acad Dermatol euros Venereol 2007; 21:251