Presentación de caso
Una mujer de 51 años fue atendida en la clínica de reumatología de este hospital debido a una secreción nasal sanguinolenta recurrente y úlceras orales.
La paciente había estado bien hasta 3,3 años antes de esta presentación, cuando desarrolló una epistaxis bilateral episódica, asociada con dolor de garganta intermitente, esputo teñido de sangre y costras nasales, sin rinorrea. Dos meses después del inicio de los síntomas, el examen realizado por un otorrinolaringólogo del Instituto de ojos y oídos de Massachusetts reveló un tabique recto, con sangrado activo de forma bilateral. Se realizó la cauterización con nitrato de plata, seguido de empaque; la sequedad nasal y el sangrado intermitente persistieron. Seis meses después del inicio de los síntomas, el examen endoscópico nasal con fibra óptica reveló costras en la nariz, inflamación de la mucosa y sinequias en la cavidad nasal izquierda, sin masas o pólipos. Se prescribió amoxicilina con clavulanato. Tres meses después, se realizó la reparación quirúrgica de la estenosis vestibular nasal bilateral, con férulas de silicona polimérica, y biopsias. El examen patológico de las muestras de biopsia mostró fragmentos superficiales de mucosa escamosa con atipia leve, hiperqueratosis, una capa granular prominente y sin evidencia de cáncer. Se administró un curso de reducción gradual de prednisona (a partir de 40 mg diarios) y una crema tópica de acetónido de triamcinolona, con una mejoría parcial. Durante el año siguiente, los síntomas persistieron a pesar de los múltiples exámenes endoscópicos nasales de fibra óptica, el desbridamiento del tejido con costra, la aplicación de antibióticos tópicos, ungüento y los enjuagues nasales.
Aproximadamente 20 meses después de la aparición de los síntomas, el examen reveló una obstrucción completa de las fosas nasales, con formación de costras y sin pólipos o secreciones; En los cultivos nasales creció flora normal. Después de un desbridamiento adicional se aplicó pomada de mupirocina. Los cultivos de seguimiento desarrollaron anaerobios moderados y pocos Staphylococcus aureus. Se prescribió un curso de 2 semanas de clindamicina.
Aproximadamente 1 año antes de esta evaluación, se desarrollaron úlceras dolorosas en la mucosa bucal y la lengua. Se administraron múltiples cursos de prednisona (hasta 40 mg al día, disminuyendo rápidamente durante un período de 1 semana), con una mejoría transitoria durante cada curso. Las pruebas para detectar anticuerpos antineutrófilos citoplasmáticos (ANCA) y anticuerpos antinucleares fueron negativas. Una radiografía de tórax y los resultados de la tomografía computarizada (TC) de los senos fueron normales. Durante los siguientes 4 meses, los episodios de epistaxis aumentaron en frecuencia, en asociación con la corteza nasal y la congestión sinusal.
Ocho meses antes de esta evaluación, en la evaluación realizada por un reumatólogo en este hospital, la paciente informó disnea, tos, fatiga, plenitud auditiva y pérdida de peso de 7 kg. El examen reveló signos vitales normales y una ulceración en la punta de la lengua; El resto del examen era normal. El nivel en sangre del anticuerpo de peroxidasa tiroidea fue de 291.0 UI por mililitro (rango de referencia, menos de 35.0), y los anticuerpos contra el ADN de doble cadena fueron positivos a una dilución de 1:40 (referencia, negativos a 1:10). El hemograma completo fue normal, al igual que los niveles de electrolitos, calcio, glucosa, proteína total, albúmina, globulina, creatina quinasa, proteína C reactiva e inmunoglobulinas (IgG, IgA e IgM); Los resultados de las pruebas de función renal y hepática y electroforesis de proteínas séricas fueron normales. Las pruebas para la sífilis (con el uso de reagina plasmática rápida), el factor reumatoide, el anticuerpo antinuclear y ANCA fueron negativos, al igual que la prueba de anticuerpos contra Ro, La, Sm y la proteína ribonuclear. Colchicina, 0,6 mg dos veces al día, se administró durante 1 semana, pero se suspendió debido a diarrea. El paciente comenzó a usar enjuague bucal con dexametasona elixir, con mejoría en las lesiones orales. La TC del tórax reveló una hernia hiatal.
Aproximadamente 5 meses antes de esta evaluación, se desarrolló un sangrado rectal intermitente. Según los informes, el examen colonoscópico realizado en otro hospital mostró dos úlceras, a 25 cm del borde anal, sin inflamación circundante. El examen patológico de las muestras de biopsia de las lesiones no mostró evidencia de ulceración o vasculitis; hubo hallazgos leves inespecíficos de criptitis aguda focal y agregados linfoides raros. La evaluación serológica para la enfermedad intestinal inflamatoria fue negativa, excepto por la presencia de anticuerpos IgG contra Saccharomyces cerevisiae (34.9 ELISA [ensayo inmunoenzimático] por mililitro; rango de referencia, menos de 17.8). Se prescribieron omeprazol, colchicina y vitamina B, sin mejoría. La paciente fue remitida a la clínica de reumatología de este hospital.
La paciente calificó el dolor de las úlceras orales en 8 en una escala de 0 a 10, con 10 que indican el dolor más grave y reportó epistaxis recurrente, hemorragia digestiva baja, fatiga, sueño no reparador, artralgias y dolor intermitente en los codos y Piernas, sin hinchazón, eritema o aumento del calor. No tenía alopecia, uveítis, erupción (malar u otra), úlceras genitales ni fístulas anales o perianales. Tenía antecedentes de tiroiditis de Hashimoto, hiperlipidemia y migrañas. Un ataque isquémico transitorio había ocurrido 3.5 años antes de esta evaluación, asociado con tinnitus en el oído derecho, entumecimiento y debilidad en el lado izquierdo y posible afasia; los síntomas se habían resuelto después de varias horas. Los medicamentos diarios incluyeron atenolol, clorhidrato de ciclobenzaprina, levotiroxina, prednisona (20 mg, disminuyendo de 40 mg durante un período de 7 días), simvastatina y aspirina (81 mg). Era alérgica al trimetoprim: sulfametoxazol, amoxicilina con clavulanato, eritromicina y tetraciclina (que había provocado erupciones cutáneas) y ciprofloxacina.
La paciente estaba casada, tenía dos hijos mayores y trabajaba en una oficina. Bebía alcohol ocasionalmente y no fumaba ni usaba drogas ilícitas. Su madre y su hermana tenían enfermedad celíaca, y su hermana también tenía esclerodermia.
En el examen, la presión arterial fue de 140/91 mm Hg y otros signos vitales eran normales. El peso fue de 76,2 kg. La orofaringe era rosada, con numerosas erosiones de las membranas mucosas de la boca, incluida la lengua, el paladar blando y la mucosa bucal interna, sin sangrado (Figura 1). El resto del examen era normal. La velocidad de sedimentación del eritrocito y el nivel de proteína C reactiva fueron normales.



Figura 1 Fotografías clínicas del paciente.
Las úlceras orales están presentes en la mucosa labial superior (Panel A), la mucosa labial inferior (Panel B) y la superficie ventral de la lengua (Panel C)
Se realizó un procedimiento diagnóstico.
DIAGNÓSTICO DIFERENCIAL
Las características más destacadas en este caso son las siguientes: mujer de mediana edad con un historial de 3 años de epistaxis, dolor de garganta, plenitud en los oídos, úlceras del tracto gastrointestinal inferior y úlceras orales multifocales dolorosas que respondieron a dosis moderadas de prednisona y glucocorticoides tópicos, específicamente acetonida de triamcinolona y dexametasona. Las muestras de biopsias de vestíbulo nasal mostraron cambios inflamatorios y reactivos no específicos, pero es importante señalar que las muestras procedían de biopsias superficiales.
Las úlceras multifocales crónicas persistentes en la cavidad oral se observan en las siguientes tres categorías generales de afecciones: infecciones, trastornos inmunitarios y trastornos ampollares autoinmunes. Algunas características de diferenciación importantes incluyen el tamaño y el aspecto de las lesiones, la participación de los sitios queratinizados frente a los no queratinizados (especialmente la encía), la curación completa de las lesiones entre brotes y la participación de la piel.
INFECCIONES VIRALES
Muchas infecciones virales (por ejemplo, enfermedad de manos, pies y boca y herpangina, causada por coxsackievirus o enterovirus) dan lugar a ulceraciones multifocales agudas en la cavidad oral que son autolimitadas; la naturaleza persistente de las úlceras de este paciente elimina la mayoría de estas. Los virus de la familia del herpesvirus establecen un estado de latencia y pueden causar úlceras orofaríngeas u orales recurrentes. El responsable más común es el virus del herpes simple (VHS). Las infecciones por VHS recurrentes se presentan con frecuencia como herpes labial y, en pacientes inmunocompetentes, como úlceras en la mucosa queratinizada de la encía, el paladar duro y el dorso de la lengua. En el huésped inmunocomprometido, puede producirse un recrudecimiento de la infección por VHS en cualquier sitio de la mucosa oral y puede parecerse a aftas orales.1 Sin embargo, las úlceras de apaciente respondieron a la prednisona, que habría exacerbado una infección por HSV.
TRASTORNOS INMUNOMEDIADOS
GRANULOMATOSIS CON POLIANGEITIS
La granulomatosis con poliangeitis (anteriormente conocida como granulomatosis de Wegener) es un trastorno vasculítico necrotizante y granulomatoso con afectación de las vías respiratorias superiores, los pulmones y los riñones; los pacientes con esta y otras vasculitis asociadas a ANCA pueden presentar úlceras orales2,3. Los pacientes con una variante de granulomatosis con poliangeitis pueden presentar una participación limitada de las vías respiratorias superiores y la cavidad oral. Sin embargo, las úlceras orales tienden a ser grandes y necróticas, y la encía típicamente muestra hiperplasia con hemorragias petequiales ("gingivitis de fresa"). 4 ANCA con especificidad para serina proteinasa 3 (más común) o mieloperoxidasa (menos común) son casi siempre positivas .3 En este caso, los hallazgos clínicos y de laboratorio (incluidos los resultados de las pruebas de función renal y las radiografías de tórax) no son compatibles con este diagnóstico.
ULCERAS AFTOSAS Y AFTAS LIKE
Las llamadas úlceras aftosas idiopáticas recurrentes son probablemente mediadas por el sistema inmunitario 5, generalmente se observan por primera vez en pacientes en la segunda y tercera décadas de la vida y tienden a aparecer durante períodos de estrés y después de un traumatismo local. Las úlceras están cubiertas por una membrana amarilla compuesta de fibrina coagulada, están rodeadas por un halo eritematoso y están confinadas a la mucosa no queratinizada, móvil (no masticatoria) (es decir, la mucosa bucal y labial y la mucosa de la lengua ventral, piso de la Boca, y paladar blando). Hay tres formas clínicas. Las aftas menores, que son las más comunes, se asocian con úlceras de menos de 1 cm en su dimensión más grande que curan en 5 a 10 días sin dejar cicatriz. Las aftas mayores se asocian con úlceras de más de 1 cm que tardan semanas o meses en curar y pueden dejar cicatriz. En las aftas herpetiformes, hay más de 10 úlceras, que miden de 0,1 a 0,5 cm, por episodio. Las úlceras en esta paciente son bastante grandes, pero no muestran evidencia de cicatrización; por lo tanto, es poco probable que representen aftas mayores.
Se observan úlceras aftosas en pacientes con trastornos sistémicos, como deficiencias hematínicas, enfermedad de Crohn, enteropatía sensible al gluten, sensibilidad a los alimentos, sensibilidad al lauril sulfato de sodio (un detergente que se encuentra en la mayoría de las pastas dentales), reacción adversa a los medicamentos, infección con la virus de inmunodeficiencia humana: síndrome de inmunodeficiencia adquirida, síndrome de PFAPA (fiebre periódica, adenopatía, faringitis y aftas), otros síndromes de aftosis compleja y enfermedad de Behçet. Un historial preciso del paciente, la eliminación de agentes sospechosos y los análisis de sangre simples descartarían la mayoría de estas afecciones. La muestra de biopsia gastrointestinal de esta paciente mostró criptitis y no granulomas; había anticuerpos contra S. cerevisiae, asociados con la enfermedad de Crohn y la enfermedad de Behçet, pero el nivel de proteína C reactiva era normal. Creo que es poco probable que sus úlceras orales estén relacionadas con la enfermedad inflamatoria intestinal.
La enfermedad de Behçet, un trastorno multisistémico inflamatorio y vasculítico, se caracteriza por úlceras aftoides orales en todos los pacientes, úlceras genitales, lesiones oculares (a menudo uveítis o vasculitis retiniana), lesiones cutáneas (por ejemplo, eritema nudoso) y, a menudo, una prueba de patología positiva. 6,7 La enfermedad tiende a afectar a hombres y mujeres turcos y japoneses jóvenes, con una fuerte asociación con el alelo HLA-B51. La afectación gastrointestinal tiende a localizarse alrededor del área ileocecal y, por lo general, respeta el recto. Este paciente no tenía una enfermedad genital u ocular, lo que hace improbable un diagnóstico de la enfermedad de Behçet.
ERITEMA MULTIFORME
El eritema multiforme ocurre en la cavidad oral como episodios agudos recurrentes de úlceras coalescentes en una mucosa eritematosa difusa (queratinizada o no queratinizada), generalmente 1 o 2 semanas después de la reactivación o recrudecimiento de la infección por VHS. Algunos casos están asociados con la exposición a conservantes de alimentos, como benzoatos.9 Las lesiones cutáneas se observan a menudo, pero no siempre, en el eritema multiforme.10 La naturaleza crónica de las úlceras de este paciente hace que este diagnóstico sea improbable.
LIQUEN PLANO
El liquen plano oral suele producir estrías blancas simétricas bilateralmente en la mucosa bucal, la mucosa de los labios, la lengua y la encía adherida. Las áreas de eritema y erosión son comunes (particularmente en la encía), al igual que las áreas de ulceración11. Sin embargo, las úlceras en el liquen plano oral casi siempre ocurren concomitantemente con lesiones reticulares y eritematosas, que estuvieron ausentes en este paciente.
ENFERMEDADES AMPOLLARES AUTOINMUNES
Por varias razones, el diagnóstico más probable para este paciente es un trastorno ampollar autoinmune, como pénfigo vulgar o pénfigo de las membranas mucosas (también conocido como pénfigoide cicatricial). Primero, el aspecto de la lesión como una membrana que recubre la mucosa es más sugestiva de una ampolla colapsada que de una úlcera verdadera, aunque puede ser difícil distinguir entre las dos. En segundo lugar, la epistaxis y la ronquera sugieren una afectación multifocal de la mucosa, que es común en las enfermedades ampollares, y las sinequias nasales sugieren penfigoide en particular. Tercero, la edad y el sexo de la paciente son típicos de los pacientes que tienen una de las dos condiciones.
El penfigoide de las membranas mucosas es un trastorno ampollar subepitelial. Los pacientes típicamente se presentan con denudación de las encías de color rojo brillante dolorosa (gingivitis descamativa), o con úlceras en el paladar.12 Hemorragias con el cepillado de dientes son comunes. Puede ocurrir ruptura de la bulla con erosión en otros sitios mucosos aunque ellos se acompañan casi siempre de compromiso gingival o de paladar o de ambos. Las cicatrices son comunes en elpenfigoide ocular pero no en el penfigoide oral y las lesiones erosivas de laringe,faringe, y mucosa nasal ocurren aproximadamente en 35% de los casos. 13 Las lesiones erosivas pueden llevar a la cicatriz, estenosis,sinequias, obstrucción y disnea. Las úlceras de esta paciente son consistentes con el diagnóstico de penfigoide de las membranas mucosas aunque no existe aparentemente compromiso gingival. Ella no tenía niveles elevados de IgG lo cual es consistente con el diagnóstico<; sin embargo, las úlceras en el colon son inusuales.
El pénfigo vulgar es un trastorno ampollar intraepitelial. La lesión comienza en la cavidad oral en hasta el 60% de los casos con lesiones que desarrollan en piel meses o años más tarde.14,15 El paladar blando y la mucosa del paladar duro están comúnmente afectados debido al trauma de la deglución. Las manifestaciones gingivales son similares a aquellas del penfigoide de las membranas mucosas. El compromiso laríngeo o faríngeo está presente en el 80% de los pacientes, costras nasales y erosiones en hasta el 60% y los oídos en hasta el 20%.16
El pénfigo vulgar es un trastorno ampollar intraepitelial. Las lesiones comienzan en la cavidad oral hasta en un 60% de los pacientes, con lesiones en la piel que se desarrollan meses o años más tarde.14,15 El paladar blando y la mucosa del paladar duro suelen estar involucrados debido a un traumatismo por tragar. Las manifestaciones gingivales son similares a las del penfigoide de la membrana mucosa. La afectación laríngea o faríngea está presente en hasta el 80% de los pacientes, la costra nasal y las erosiones se encuentran en hasta el 60% y las orejas en el 20% .16 Las úlceras orales de esta paciente son compatibles con este diagnóstico. En un paciente con solo afectación de la mucosa oral, no esperaría que los niveles de IgG estén elevados; Las úlceras en el colon son inusuales en el pénfigo vulgar.
El síndrome multiorgánico autoinmune paraneoplásico (pénfigo paraneoplásico), la enfermedad de IgA lineal, el lupus eritematoso y la epidermólisis ampollosa son otras enfermedades ampollosas autoinmunes; Los pacientes pueden presentar úlceras en la cavidad oral y otras membranas mucosas, pero siempre con afectación concomitante de la piel, anomalías serológicas o ambas. Estas enfermedades son poco probables en este caso.
Es difícil distinguir clínicamente entre penfigoide de membrana mucosa oral y pénfigo vulgar oral; En cada afección, hay afectación de la mucosa laríngea y nasal, como se ve en este paciente. La presencia de sinequias nasales en esta paciente es más sugestiva de penfigoide de membrana mucosa.
El procedimiento de diagnóstico debe ser otra biopsia de mucosa perilesional, clínicamente no comprometida, tanto para el examen histopatológico de rutina como para estudios de inmunofluorescencia directa de la muestra.
SERVICIO DE REUMATOLOGÍA: El servicio de reumatología consideró la enfermedad de Behçet, otras formas de vasculitis sistémica y varias afecciones autoinmunes, incluido el lupus. Debido a que algunas de las lesiones orales tenían la apariencia de una membrana que cubría la úlcera debajo de una ampolla colapsada, en lugar de una úlcera aftosa clásica, sospechamos un trastorno ampolloso autoinmune.
DIAGNOSTICO CLÍNICO PRESUNTIVO
TRASTORNO AMPOLLAR AUTOINMUNE, PÉNFIGO VULGAR O PENFIGOIDE DE MEMBRANAS MUCOSAS.
DISCUSION PATOLOGICA
Los primeros procedimientos de diagnóstico fueron biopsias del vestíbulo nasal. Los especímenes mostraron liquen simple crónico, que es un patrón histológico inespecífico que se puede observar en el trauma mecánico en curso. El desencadenante inicial puede ser una dermatitis eccematosa u otro proceso subyacente. No se observó evidencia diagnóstica de un trastorno ampolloso autoinmune. Sin embargo, los estudios de inmunofluorescencia directa habrían sido útiles para una evaluación adicional. La biopsia fue demasiado superficial para evaluar completamente la cicatrización. La muestra de biopsia rectal también mostró hallazgos inespecíficos.
El procedimiento diagnóstico consistió en biopsias de mucosa oral. Las muestras se enviaron para el procesamiento histológico de rutina y la inmunofluorescencia directa. El examen de rutina mostró ulceración epidérmica y hendiduras subepiteliales. El infiltrado inflamatorio consistió en linfocitos, neutrófilos y eosinófilos dispersos (Figura 2).

Figura 2A, 2B y 2C Figura 2 Muestras de biopsia de la mucosa oral.
El panel A (hematoxilina y eosina) muestra una ulceración parcial de la mucosa. El panel B (hematoxilina y eosina) muestra formación de hendidura subepitelial y un infiltrado linfocítico asociado que contiene neutrófilos y eosinófilos. El panel C (hematoxilina y eosina) muestra eosinófilos concentrados focalmente alrededor de la zona de la membrana basal . El panel D muestra tinción lineal para IgG en la membrana basal con inmunofluorescencia directa.
Estos cambios abren un amplio diagnóstico diferencial (tabla 1).


Tabla 1 Diagnóstico diferencial histológico.
Las úlceras aftosas recurrentes y la enfermedad de Behçet suelen tener características histológicas inespecíficas y no muestran hendiduras subepiteliales. La infección por herpesvirus muestra cambios nucleares virales en los queratinocitos, que no se observaron aquí. Debido a la presencia de formación de hendidura subepitelial, el diagnóstico diferencial incluye los siguientes dos grupos principales de afecciones: afecciones que ocasionan mucositis de interfase (liquen plano, eritema multiforme y lupus eritematoso) y los trastornos ampollosos autoinmunitarios (pénfigo vulgar, penfigoide bulloso, penfigoide de las membranas mucosas y pénfigo paraneoplásico). Aunque el examen histológico de rutina puede proporcionar pistas sutiles para permitir que se haga una distinción entre los dos grupos, la inmunofluorescencia directa es esencial para un diagnóstico preciso (Figura 3).


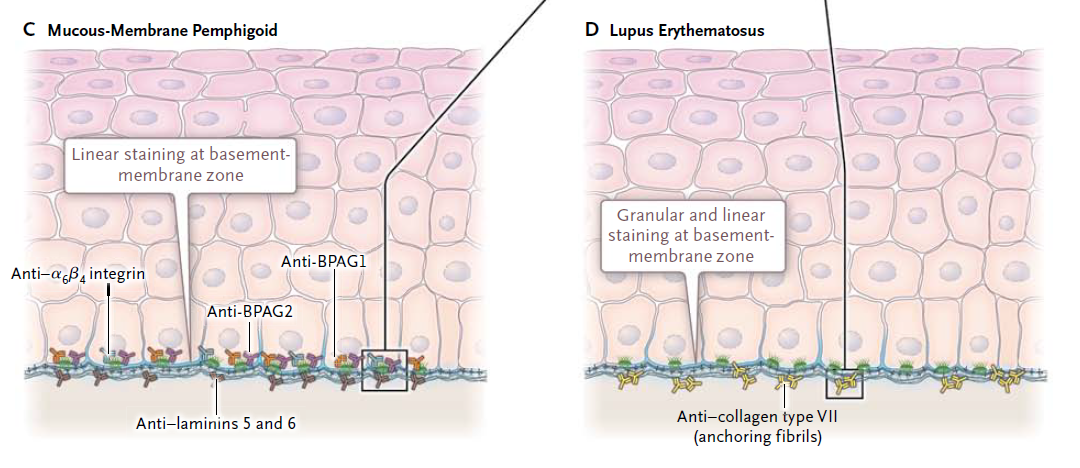
Figura 3 Mecanismos patogénicos y deposición de anticuerpos en los trastornos de ampollas.
En el pénfigo (Panel A), los autoanticuerpos atacan antígenos desmosomales (desmoglein 1 y desmoglein 3). Los desmosomas conectan los queratinocitos epiteliales, lo que da como resultado la tinción intercelular en un patrón de red en inmunofluorescencia directa y acantólisis con formación de vesículas intraepiteliales. En el penfigoide bulloso (Panel B), los autoanticuerpos se unen a los antígenos hemidesmosomales (antígenos del penfigoide bulloso 1 y 2 [BPAG1 y BPAG2]). Los hemidesmosomas median la unión de los queratinocitos a la membrana basal. Por lo tanto, se observan tinciones lineales en la membrana basal con inmunofluorescencia directa y una ampolla subepitelial. En el penfigoide de membrana mucosa (Panel C), los anticuerpos se detectan con mayor frecuencia contra BPAG2 y la cadena beta de la integrina α6 β4, que se extiende desde los hemidesmosomas hasta la lámina lúcida. Además, se observan anticuerpos dirigidos contra la laminina 5 (epiligrina, un componente de la lámina lúcida) y contra BPAG1 y laminina 6. El subtipo anti-laminina 5 muestra una asociación con condiciones malignas internas. Se observan tinciones lineales en la membrana basal con inmunofluorescencia directa y una ampolla subepitelial, similares a las observadas en el penfigoide bulloso. En el lupus eritematoso (Panel D), los anticuerpos contra el colágeno tipo VII dentro de las fibrillas de anclaje conducen a una forma granular (y algunas veces lineal)
MUCOSITIS DE INTERFASE
La mucositis de interfase es un proceso inflamatorio de la unión epitelial-subepitelial y conduce a la degeneración vacuolar de los queratinocitos basales. Los queratinocitos apoptóticos (cuerpos coloides) se ven a menudo. Estos cambios degenerativos pueden conducir a la formación de fisuras y ulceración, como se ve en la muestra de biopsia de esta paciente; sin embargo, en este espécimen no se observaron queratinocitos apoptóticos. Además, los eosinófilos alrededor de la zona de la membrana basal, como se ve en este caso, no son una característica típica de la mucositis de interfase. La inmunofluorescencia directa revela hallazgos inespecíficos en el liquen plano (Tabla 1). Las reacciones a los medicamentos a menudo se asocian con cambios en la interfase que involucran eosinófilos prominentes y no hay hallazgos sobre la inmunofluorescencia directa. El lupus eritematoso es probablemente la entidad líder en este grupo en el diagnóstico diferencial para este caso, pero los eosinófilos generalmente no están presentes.
TRASTORNOS AMPOLLOSOS AUTOINMUNES
El grupo de los pénfigos y los penfigoides son las consideraciones diagnósticas más importantes en este caso. Los trastornos del grupo del pénfigo muestran acantólisis y formación de vesículas intraepiteliales debido a la unión de autoanticuerpos a antígenos en los desmosomas, que proporcionan unión entre los queratinocitos.17 En este caso, la hendidura es subepitelial, lo que hace improbable el pénfigo.
Los trastornos del grupo de los penfigoides son causados por anticuerpos contra las proteínas de la membrana basal, lo que lleva a hendidura subepitelial y la formación de ampollas, como se ve en este caso (Tabla 1 y Figura 2B). En el penfigoide bulloso, los antígenos principales se encuentran dentro de los hemidesmosomas que median la unión de los queratinocitos a la membrana basal.18
El penfigoide de membranas mucosas es una enfermedad heterogénea18,19 que involucra autoanticuerpos que se unen a diversos antígenos hemidesmosomales y antígenos en la lámina lúcida de la membrana basal (más comúnmente antígeno de pemphigoid bulloso 2 [BPAG2], pero también integrina α6 β4, lamininas 5 y 6, y otros) (Tabla 1 y Figura 3) .20 Similar al penfigoide bulloso, esto se traduce en tinción lineal en la membrana basal con inmunofluorescencia directa, comúnmente para IgG y complemento (C3). La presencia de eosinófilos dispersos alrededor de la zona de la membrana basal, como se ve en este caso, es una característica típica tanto del penfigoide bulloso como del penfigoide de la membrana mucosa.
En un sustrato de la piel de esta paciente, la inmunofluorescencia directa y la posterior incubación con anticuerpos conjugados con fluoresceína contra inmunoglobulinas y C3 mostraron tinción lineal para IgG y tinción lineal y parcialmente granular para C3 en la membrana basal. Estos hallazgos se observan tanto en el penfigoide de la membrana mucosa como en el penfigoide bulloso (Figura 2D).
La distinción entre penfigoide bulloso y penfigoide de membrana mucosa no se puede hacer sobre la base de la histología y la inmunofluorescencia directa solo. Es necesaria la correlación clínica (o caracterización serológica de los autoanticuerpos). En vista de las características clínicas de este caso, los hallazgos de la biopsia son consistentes con penfigoide de la membrana mucosa.
REUMATOLGIA: Según el consenso internacional de 2002, el penfigoide de la membrana mucosa ahora incluye las entidades clínicas anteriores: el penfigoide cicatricial, la epidermólisis ampollosa dominante de la membrana mucosa y la dermatosis ampollosa de IgA lineal.21 La gravedad de la enfermedad varía, pero el tratamiento Los casos resistentes son comunes. Los síntomas clínicamente significativos e incluso la muerte pueden resultar del proceso de cicatrización asociado con esta afección o de infecciones que resultan de ulceraciones extensas. Los efectos secundarios asociados con el tratamiento son frecuentes y, por lo tanto, el régimen seleccionado debe lograr un equilibrio entre la prevención de las complicaciones relacionadas con la enfermedad y la prevención de los efectos adversos graves de la terapia.
No dirigimos el tratamiento inicial de este paciente, pero ella había mostrado una excelente respuesta a los glucocorticoides en dosis altas, comenzando con prednisona a 60 mg por día. Desafortunadamente, durante los siguientes 2 meses, no pudimos reducir la dosis de prednisona por debajo de 15 a 20 mg por día sin la recurrencia de lesiones orales dolorosas. Al mismo tiempo, se desarrollaron múltiples efectos secundarios en asociación con los glucocorticoides, en particular insomnio, una facies cushingoidea y un aumento sustancial de peso. La adición de metotrexato (25 mg por semana), seguida de un inhibidor del factor de necrosis tumoral α (etanercept, 50 mg por semana), no proporcionó ningún control adicional de la enfermedad y no tuvo un efecto de ahorro de glucocorticoides.
RITUXIMAB EN EL PENFIGOIDE DE LAS MEMBRANAS MUCOSAS
Aunque la evidencia anecdótica habría respaldado los ensayos de ciclofosfamida, micofenolato mofetilo, inmunoglobulina intravenosa y otros agentes, se eligió tratar a este paciente con rituximab (1 g por vía intravenosa dos veces, separados por 15 días). Nuestro fundamento para el agotamiento de las células B por medio de rituximab fueron los informes sobre el sorprendente éxito del tratamiento con el agotamiento de las células B para casos refractarios de pénfigo vulgar y pénfigo foliáceo y un pequeño número de casos de pénfigoide de membranas mucosas2-22. , se ha observado el autoanticuerpo primario tanto en el pénfigo vulgar como en el pénfigo foliáceo de la subclase IgG4, y se ha observado una rápida eficacia de este enfoque de tratamiento en pacientes con enfermedad relacionada con IgG4 resistente a los glucocorticoides25,26. El penfigoide de membrana mucosa está asociado con una variedad de anticuerpos, pero, según nuestro conocimiento, no se han realizado informes sistemáticos de las subclases de IgG específicas involucradas en estas condiciones.
La paciente está aquí con nosotros hoy. ¿Podrías decirnos cómo te va ahora?
LA PACIENTE: Había estado sintiendo síntomas muy incómodos durante algún tiempo, lo que afectó significativamente mi calidad de vida. En mi búsqueda de respuestas, ocasionalmente me trataron con una actitud desdeñosa y me hicieron sentir que mis problemas realmente no eran tan malos. Cuando llegué al Hospital General de Massachusetts, me trataron con profesionalismo, atención y preocupación. Mis síntomas fueron validados, y parecía haber una verdadera urgencia para tratar de averiguar exactamente qué estaba mal. No puedo expresar lo importante que me hicieron sentir. Mis preguntas e inquietudes fueron respondidas con prontitud, sin importar con quién hablaba: el personal de secretaría, los asistentes médicos y, por supuesto, los proveedores. Estoy abrumado por la dignidad y el respeto que se me ha mostrado.
Un mes después de mi segunda infusión de rituximab, ya estaba volviendo a ser yo misma. Fue un viaje muy largo llegar hasta aquí. Estaba absolutamente desgraciada, tenía mucho dolor. Solo ahora que me siento tan bien puedo pensar, Dios mío, eso fue realmente miserable. No pude ni beber agua de una pajita en un momento dado. Ahora puedo beber jugo nuevamente, incluso jugo de naranja y jugo de tomate. Ahora que estoy de este lado, estoy muy agradecida.
REUMATOLOGÍA: Todos hemos estado extremadamente gratificados por la respuesta de la paciente al tratamiento. Después de recibir las infusiones de rituximab, disminuimos la dosis de prednisona de manera constante y suspendimos los glucocorticoides durante un período de 3 meses. Durante ese tiempo, la paciente tuvo solo una úlcera leve, que desapareció después de 2 días. Anteriormente, no había podido reducir la prednisona por debajo de 10 mg por día, a pesar de los otros medicamentos inmunosupresores concurrentes. Permaneció completamente sin prednisona durante 5 meses, hasta que las células B periféricas comenzaron a reconstituirse, por lo que comenzó a tener ulceraciones orales recurrentes. Recientemente la tratamos nuevamente con un segundo tratamiento de rituximab. Ella sigue teniendo sangre ocasional en las heces, pero en general se mantiene bien y no requiere prednisona.
DIAGNOSTICO ANATOMICO
PENFIGOIDE DE MEMBRANAS MUCOSAS.
Traducción de:
A 51-Year-Old Woman with Epistaxis and Oral Mucosal Ulcers
Sook-Bin Woo, D.M.D., John H. Stone, M.D., M.P.H., and Stefan Kraft, M.D.
N Engl J Med 2013; 369:265-274July 18, 2013DOI: 10.1056/NEJMcpc1209275
Referencias
1
Woo SB, Lee SF. Oral recrudescent herpes simplex virus infection. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1997;83:239-243
CrossRef | Web of Science | Medline
.
2
Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum 1990;33:1101-1107
CrossRef | Web of Science | Medline
.
3
Falk RJ, Gross WL, Guillevin L, et al. Granulomatosis with polyangiitis (Wegener's): an alternative name for Wegener's granulomatosis. Arthritis Rheum 2011;63:863-864
CrossRef | Web of Science | Medline
.
4
Stewart C, Cohen D, Bhattacharyya I, et al. Oral manifestations of Wegener's granulomatosis: a report of three cases and a literature review. J Am Dent Assoc 2007;138:338-348
Web of Science | Medline
.
5
Jurge S, Kuffer R, Scully C, Porter SR. Mucosal disease series. Number VI. Recurrent aphthous stomatitis. Oral Dis 2006;12:1-21
CrossRef | Web of Science | Medline
.
6
International Study Group for Behcet's Disease. Criteria for diagnosis of Behçet's disease. Lancet 1990;335:1078-1080
Web of Science | Medline
.
7
Mendes D, Correia M, Barbedo M, et al. Behçet's disease -- a contemporary review. J Autoimmun 2009;32:178-188
CrossRef | Web of Science | Medline
.
8
Ayangco L, Rogers RS III. Oral manifestations of erythema multiforme. Dermatol Clin 2003;21:195-205
CrossRef | Web of Science | Medline
.
9
Lewis MA, Lamey PJ, Forsyth A, Gall J. Recurrent erythema multiforme: a possible role of foodstuffs. Br Dent J 1989;166:371-373
CrossRef | Web of Science | Medline
.
10
Bean SF, Quezada RK. Recurrent oral erythema multiforme: clinical experience with 11 patients. JAMA 1983;249: 2810-2812
CrossRef | Web of Science | Medline
.
11
lcully C, Carrozzo M. Oral mucosal disease: lichen planus. Br J Oral Maxillofac Surg 2008;46:15-21
CrossRef | Web of Science | Medline
.
12
Chan LS. Ocular and oral mucous membrane pemphigoid (cicatricial pemphigoid). Clin Dermatol 2012;30:34-37
CrossRef | Web of Science | Medline
.
13
Alexandre M, Brette MD, Pascal F, et al. A prospective study of upper aerodigestive tract manifestations of mucous membrane pemphigoid. Medicine (Baltimore) 2006;85:239-252
CrossRef | Web of Science | Medline
.
14
Scully C, Paes De Almeida O, Porter SR, Gilkes JJ. Pemphigus vulgaris: the manifestations and long-term management of 55 patients with oral lesions. Br J Dermatol 1999;140:84-89
CrossRef | Web of Science | Medline
.
15
Zakka LR, Reche P, Ahmed AR. Role of MHC class II genes in the pathogenesis of pemphigoid. Autoimmun Rev 2011;11:40-47
CrossRef | Web of Science | Medline
.
16
Mignogna MD, Fortuna G, Leuci S, Ruoppo E. Oropharyngeal pemphigus vulgaris and clinical remission: a long-term, longitudinal study. Am J Clin Dermatol 2010;11:137-145
CrossRef | Web of Science | Medline
.
17
Bystryn JC, Rudolph JL. Pemphigus. Lancet 2005;366:61-73
CrossRef | Web of Science | Medline
.
18
Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity 2012;45:55-70
CrossRef | Web of Science | Medline
.
19
Bruch-Gerharz D, Hertl M, Ruzicka T. Mucous membrane pemphigoid: clinical aspects, immunopathological features and therapy. Eur J Dermatol 2007;17:191-200
Web of Science | Medline
.
20
Egan CA, Lazarova Z, Darling TN, Yee C, Yancey KB. Anti-epiligrin cicatricial pemphigoid: clinical findings, immunopathogenesis, and significant associations. Medicine (Baltimore) 2003;82:177-186
CrossRef | Medline
.
21
Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol 2002;138:370-379
CrossRef | Web of Science | Medline
.
22
Joly P, Mouquet H, Roujeau JC, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med 2007;357:545-552
Free Full Text | Web of Science | Medline
.
23
Kasperkiewicz M, Shimanovich I, Ludwig RJ, Rose C, Zillikens D, Schmidt E. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol 2011;65:552-558
CrossRef | Web of Science | Medline
.
24
Leshem YA, Hodak E, David M, Anhalt GJ, Mimouni D. Successful treatment of pemphigus with biweekly 1-g infusions of rituximab: a retrospective study of 47 patients. J Am Acad Dermatol 2013;68:404-411
CrossRef | Web of Science | Medline
.
25
Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum 2010;62:1755-1762
CrossRef | Web of Science | Medline
.
26
Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore) 2012;91:57-66
CrossRef | Web of Science | Medline