Un hombre de 29 años fue ingresado en este hospital debido a anemia e ictericia.
El paciente había estado en su salud habitual hasta 4 días antes del ingreso, cuando comenzó a presentar aumento de la fatiga, malestar general, dolor de cabeza, molestias testiculares intermitentes, se notó los ojos amarillentos, la orina oscura, y comenzó con náuseas, y dolores corporales difusos. Un dolor crónico que presentaba en las piernas empeoró. No había fiebre, erupción, hematuria, disuria, melena ni síntomas neurológicos. El día de la admisión, llegó al servicio de urgencias de este hospital.
El paciente había estado bien en general, con asma intermitente, acné y dolor crónico en las piernas hasta 4 meses antes, cuando viajó desde un área urbana de Nueva Inglaterra a la casa de su familia en el norte de África y se quedó allí por 3 meses. Mientras estuvo allí, apareció una tos prolongada productiva con esputo verde, asociada con fatiga, fiebre subjetiva, escalofríos, sudores nocturnos profusos y disnea intermitente en reposo, y refirió que tuvo una pérdida de peso de 9.1 kg. Diez días después de su regreso a Nueva Inglaterra, y 20 días antes de esta admisión, se lo vio en clínica médica de este hospital.
En la evaluación en ese momento, el paciente parecía estar bien. El peso era de 92,7 kg, el índice de masa corporal de 23,6 y el pulso de 101 pulsaciones por minuto; La presión sanguínea y las respiración eran normales. La evaluación reveló acné leve, cornetes nasales inflamados, pálidos y con secreciones claras, agrandamiento de amígdalas leve, crepitantes inspiratorios en la base pulmonar derecha, sibilancias espiratorias en ambos vértices pulmonares, y cicatrices quirúrgicas en la pierna derecha. El nivel de plaquetas y los resultados de la electroforesis de hemoglobina fueron normales y las pruebas para el virus de inmunodeficiencia humana (VIH) fueron negativas; otros resultados de la prueba se muestran en la Tabla 1. Una radiografía de tórax fue normal. Una prueba cutánea de tuberculina de Mantoux fue negativa. Se recomendó al paciente que continuara con la administración de fluticasona y albuterol mediante inhaladores, que habían sido prescritos como tratamiento para el asma. Regresó a su casa. El día de la admisión, llamó a la oficina de su médico y se le recomendó ir al departamento de emergencias para una evaluación. Posteriormente ingresó en el hospital.

Tabla 1. Datos de laboratorio.
El paciente tenía antecedentes de fracturas de la pierna derecha después de un accidente automovilístico 3 años antes, lo que requirió la fijación interna de la tibia por falta de unión, así como injertos de piel y hueso; tenía dolor crónico persistente en la pierna e hinchazón venosa. Un año antes de esta admisión, después de la cirugía tibial, el hematocrito disminuyó a 34.4%, con un volumen corpuscular medio de 77 fl (rango normal, 80 a 100); La anemia se resolvió en 4 meses. Otros medicamentos incluyeron ginkgo biloba, un multivitamínico y parches de nicotina para dejar de fumar. No tenía alergias conocidas a la medicación.
El paciente había emigrado del norte de África a los Estados Unidos 9 años antes. Era estudiante, fumaba marihuana y cigarrillos, y no reportó el uso actual de alcohol o drogas por vía intravenosa. Era sexualmente activo con su novia. Mientras el paciente visitaba a su familia en el norte de África, su hermano de 14 años murió de una enfermedad no especificada. Durante ese viaje, el paciente estuvo expuesto a ovejas y bebió leche de vaca sin pasteurizar. Su madre tenía osteopenia; su padre y otros cuatro hermanos estaban sanos.
En el examen, la presión arterial fue de 123/85 mm Hg y el pulso 115 latidos por minuto; La temperatura, las respiraciones y la saturación de oxígeno fueron normales. El paciente parecía cansado, con ictericia conjuntival y en piel. El segundo ruido cardiaco era fuerte, sin soplos. El abdomen era blando, con un dolorimiento difuso que era mayor en los cuadrantes superior derecho e izquierdo; los testículos eran difusamente sensibles, sin masas. Se observó sensibilidad leve sobre la rodilla derecha y la cesta tibial anterior, con cicatrices quirúrgicas curadas; el resto del examen era normal.
El recuento de plaquetas, el recuento diferencial de glóbulos blancos y los índices de glóbulos rojos (volumen corpuscular medio y concentración media de hemoglobina corpuscular) fueron normales, al igual que los resultados de las pruebas de coagulación y los niveles sanguíneos de electrolitos, glucosa, calcio, magnesio, proteína total, albúmina, globulina, fosfatasa alcalina, alanina y aspartato aminotransferasas, lipasa, tirotropina, folato y vitamina B12. Los frotis de sangre periférica gruesos y delgados y las pruebas rápidas para el antígeno de la malaria, los anticuerpos contra el VIH y los anticuerpos heterófilos fueron negativos; otros resultados de la prueba se muestran en la Tabla 1. Una prueba de antiglobulina directa (prueba de Coombs) fue negativa. Se obtuvieron cultivos de la sangre. Los resultados del análisis de orina fueron normales y las heces fueron negativas para el guayaco. Un electrocardiograma fue normal. Una radiografía de tórax mostró pulmones claros. Se administraron analgesia narcótica, ondansetrón, dalteparina, suplementos de fósforo y líquidos intravenosos. El examen toxicológico de la orina reveló la presencia de opiáceos y cannabinoides.
Durante los siguientes 2 días, la ictericia mejoró gradualmente. Los hemocultivos permanecieron estériles. Los resultados de las pruebas adicionales se muestran en la Tabla 1. Un examen de ultrasonido del abdomen reveló una esteatosis hepática leve; La ecografía de testículos fue normal.
En el cuarto día, se realizó una prueba diagnóstica.
DIAGNÓSTICO DIFERENCIAL
Un hombre de 29 años del norte de África con antecedentes de asma, acné y dolor crónico en las piernas ingresó en este hospital después de 4 días de fatiga, malestar general, dolor de cabeza, dolor corporal difuso, ojos amarillentos, orina oscura, malestar testicular y dolor en las piernas. Se encontró que estaba marcadamente anémico.
Quizás la parte más difícil de este caso es dar sentido a todos los síntomas aparentemente no relacionados y los hallazgos clínicos que se descubrieron durante la presentación y decidir cuáles son los signos y síntomas primarios de un diagnóstico presuntivo y que están simplemente asociados, o incluso no relacionados con, este diagnóstico Aunque el paciente estaba relativamente sano, un reciente viaje a África lo llevó a una larga enfermedad, con tos prolongada, fiebre, sudores nocturnos y una pérdida de peso de 9.1 kg. A pesar de la aparente falta de tratamiento, se recuperó. Sin embargo, pocos días después de regresar a los Estados Unidos, comenzó a sentirse mal de nuevo, lo que finalmente condujo a la admisión en el hospital. Nos queda preguntarnos si estos dos episodios de enfermedad están relacionados.
Hay varias características de este caso que son potencialmente importantes pero que no se correlacionan fácilmente con la presentación del paciente y, por lo tanto, nos dejan preguntándonos sobre su importancia clínica. Específicamente, la fiebre, la tos y la pérdida de peso se desarrollaron mientras el paciente estaba visitando el norte de África, donde se encontró con un hermano moribundo, estuvo expuesto a las ovejas y bebió leche sin pasteurizar. Además, nos enteramos de que su historial médico es notable por un episodio previo de anemia aguda que ocurrió después de una cirugía. En última instancia, este joven se presenta con orina oscura y signos y síntomas de marcada anemia, y debemos buscar un diagnóstico unificador para estos hallazgos.
ANEMIA HEMOLÍTICA
Revisamos los datos de laboratorio obtenidos en la admisión y los comparamos con los datos obtenidos durante una visita de atención primaria 20 días antes de la admisión. Hubo una caída rápida en el nivel de hemoglobina del paciente, de 14.8 g por decilitro antes del ingreso a 10.2 g por decilitro al ingreso. Tal disminución se explica ya sea por la pérdida de sangre o por destrucción de glóbulos rojos (es decir, la hemólisis). Durante los 2 días posteriores al ingreso, la hemoglobina disminuyó constantemente a un nadir de 9,3 g por decilitro. El paciente tenía ictericia cutánea y conjuntivas ictéricas, niveles marcadamente elevados de lactato deshidrogenasa sérica y bilirrubina no conjugada en asociación con una función hepática normal, un nivel de haptoglobina casi indetectable y ninguna fuente aparente de sangrado. Teniendo en cuenta todos estos factores, podemos estar relativamente seguros de que un proceso hemolítico es responsable de la caída aguda en el nivel de hemoglobina. Aunque me gustaría ver una reticulocitosis más robusta en respuesta a la destrucción de los glóbulos rojos, el hecho de que el porcentaje de reticulocitos en el ingreso (3,3%) no sea adecuado para este nivel de anemia no debería disuadirnos de considerar la hemólisis como la razón de esta anemia aguda del paciente. Con el tiempo, se esperaría una respuesta más sólida si el paciente tuviera una médula ósea en funcionamiento y los componentes básicos para producir eritrocitos nuevos e inmaduros.
GENES, CONDUCTAS O MALA SUERTE
Este paciente es muy joven para estar tan enfermo. En un paciente relativamente joven, muchas enfermedades se pueden agrupar fácilmente en una de las siguientes tres categorías generales: enfermedades causadas por herencia (genes), aquellas causadas por exposiciones que a menudo se deben a un comportamiento o conducta particular y aquellas que son consecuencia de la vida cotidiana (mala suerte). Aunque simple, este enfoque puede ayudarnos a catalogar rápidamente los tipos de trastornos que pueden ser responsables de la presentación de este joven. El diagnóstico de anemia hemolítica no es difícil de realizar; sin embargo, encontrar una causa para el proceso hemolítico puede resultar más desafiante.
Obteniendo una historia familiar completa con atención particular a las enfermedades hereditarias, un detallado historial del paciente que incluya antecedentes de enfermedades y exposiciones (viajes y consumo de drogas), y un fotis de sangre periférica debe conducir a la causa correcta en la mayoría de los casos de hemólisis. Es costumbre categorizar a la hemólisis como un proceso extravascular o un proceso intravascular, dependiendo de si la destrucción de las células rojas ocurre fuera o dentro del sistema retículo endotelial.. Sin embargo, para evitar algunos de las trampas de este enfoque, se ha sugerido que la anemia hemolítica se categorice como un trastorno heredado o adquirido del glóbulo rojo.1 Encuentro este enfoque simple y fácil de seguir; este paciente podría tener un gen que provoca un trastorno hemolítico hereditario de los glóbulos rojos. Alternativamente, pudo haber adquirido un desorden a través de una exposición (por ejemplo, ambiental, laboral, o infeccioso) o a través del desarrollo de una condición que resulta en hemólisis (por ejemplo, una condición maligna o una picadura de insecto).
CAUSAS ADQUIRIDAS DE HEMÓLISIS
Al considerar las posibles causas adquiridas de hemólisis, debemos intentar descartar hiperesplenismo, hemólisis mediada por el sistema inmunitario, microangiopatía, infección y algunos otros trastornos poco comunes, como la hemoglobinuria paroxística nocturna y la hemoglobinuria paroxística por frío (Tabla 2). Aunque aún no hemos examinado un frotis de sangre periférica, se nos dice que el paciente tenía una función hepática normal y, según las imágenes y el examen, no tenía esplenomegalia, lo que hace improbable el secuestro y la hemólisis mediada por el sistema reticuloendotelial. Una prueba de anticuerpos directa no fue reactiva y no se informaron esferocitos, por lo que la hemólisis mediada por el sistema inmune también es poco probable. El paciente tenía un recuento plaquetario normal y resultados normales de los estudios de coagulación, y no se observaron esquistocitos en el recuento sanguíneo completo. Estos hallazgos esencialmente descartan un proceso hemolítico microangiopático, como la púrpura trombocitopénica trombótica o la coagulación intravascular diseminada. La malaria es una posibilidad, dado su reciente viaje a un área en la que la enfermedad es endémica, pero los frotis gruesos y delgados de sangre periférica y una prueba rápida para el antígeno de la malaria fueron negativos. Sobre la base de pruebas serológicas negativas, no son probables otras infecciones (por ejemplo, VIH y virus de Epstein-Barr). Aunque no se ha descartado definitivamente la tuberculosis, es poco probable, dada la prueba de Mantoux negativa.

Table 2 Causas de anemia hemolítica.
CAUSAS HEREDITARIAS DE HEMÓLISIS
El paciente es joven y no hay pruebas que respalden el diagnóstico de un proceso hemolítico adquirido, lo que constituye un fuerte argumento a favor de una causa hereditaria de anemia hemolítica. Las anemias hemolíticas hereditarias se pueden clasificar en función de la anomalía de los glóbulos rojos que es responsable de su destrucción; por lo tanto, la hemólisis hereditaria puede ser causada por un defecto de membrana, un defecto enzimático o defectos de hemoglobina intrínsecos a los glóbulos rojos. De los defectos de la membrana que explican la mayoría de las causas hereditarias de la anemia hemolítica, tanto la esferocitosis hereditaria como la eliptocitosis hereditaria son trastornos autosómicos dominantes; a nuestro entender, este paciente no tiene otros miembros de la familia afectados, lo que hace que estos trastornos sean poco probables. Además, los índices de glóbulos rojos, particularmente la concentración de hemoglobina corpuscular media, tienden a ser anormales en ambas condiciones. Del mismo modo, podemos descartar muchas de las hemoglobinopatías, incluidas la anemia falciforme y la talasemia, sobre la base del volumen corpuscular medio normal de este paciente y la ausencia de anemia en el curso de su vida.
Nos quedan por considerar los trastornos hemolíticos hereditarios que se deben a un defecto enzimático. De estos, la deficiencia de piruvato quinasa es un trastorno hereditario autosómico que afecta predominantemente a personas de ascendencia del norte de Europa. En contraste, la deficiencia de glucosa-6-fosfato deshidrogenasa (G6PD) es un trastorno recesivo ligado a X que se encuentra principalmente en hombres de ascendencia asiática, africana, mediterránea y del Medio Oriente, y por lo tanto parece ser una posibilidad clara en este paciente. A diferencia de las otras anemias hemolíticas hereditarias, la deficiencia de G6PD suele ser una enfermedad autolimitada que se presenta poco después de un ataque oxidativo a los glóbulos rojos, como infección, exposición a medicamentos o ingestión de habas. Los signos y síntomas de hemólisis se desarrollan en pacientes con esta deficiencia de enzimas, pero los pacientes suelen tener índices de eritrocitos normales y pocas anomalías de laboratorio. Este paciente tenía índices normales de glóbulos rojos, y su condición mejoró rápidamente sin intervención terapéutica, factores que apoyan el diagnóstico de deficiencia de G6PD.
La enzima G6PD tiene un papel crítico en la reducción de glutatión y NADP en la vía de las pentosas fosfato dentro de los eritrocitos. Tanto el glutatión como el NADP son necesarios para defenderse contra el estrés oxidativo celular. Bajo condiciones de estrés oxidativo, la hemoglobina se desnaturaliza y los glóbulos rojos se vuelven susceptibles a una hemólisis inminente en personas con deficiencia de estas enzimas. Aunque en este caso no está claro cuál pudo haber sido el posible insulto o el factor de estrés, algunos datos sutiles pero notables nos llevan a creer que la deficiencia de G6PD podría haber causado hemólisis en este paciente. Estas pistas incluyen su ascendencia (norteafricana y mediterránea), la anemia hemolítica autolimitada que se resolvió rápidamente después del ingreso hospitalario, la leve disminución de los niveles de hierro durante el transcurso de unos días (lo que sugiere una reticulocitosis enérgica) y el episodio anterior de anemia después de una cirugía. Una extensa lista de medicamentos que deben evitar los pacientes con deficiencia de G6PD no incluye inhaladores ni parches de nicotina, pero los efectos del ginkgo biloba en este trastorno no están claros.2 La relación entre el acné y la G6PD ha sido debatida durante mucho tiempo, y un informe reciente No sugirió ninguna asociación definitiva.
No está claro qué evento o exposición particular provocó la crisis hemolítica del paciente y la consiguiente presentación clínica. Sin embargo, no tenemos explicación para una serie de detalles, incluyendo su reciente enfermedad prolongada mientras visitaba su país; la pérdida de peso, la tos y la fiebre; y si su exposición a las ovejas, su ingestión de leche no pasteurizada y el hallazgo de esteatosis hepática en el examen de ultrasonido están relacionados. Sin embargo, el diagnóstico de deficiencia de G6PD se puede realizar sobre la base de una historia bien documentada, evidencia de hemólisis, un frotis de sangre periférica que muestra cuerpos de Heinz (eritrocitos con hemoglobina desnaturalizada) y “células mordidas” (bite cells), y la medición del nivel de actividad de la G6PD. Es importante recordar que el nivel de actividad de la enzima puede permanecer normal durante un episodio hemolítico agudo porque solo se analizan las células rojas más jóvenes no hemolizadas y recién producidas. Si la deficiencia de G6PD sigue siendo una preocupación o sospecha después de que se mide la actividad normal, el ensayo debe repetirse unas semanas más tarde, una vez que la hemólisis ha cesado y las células de todas las edades están nuevamente presentes.4 La enfermedad es autolimitada y el tratamiento está dirigido a detener la hemólisis identificando, eliminando o tratando el agente responsable del aumento del estrés oxidativo. Los objetivos terapéuticos están dirigidos a mantener un nivel de hemoglobina que sea suficiente para la estabilidad hemodinámica, y las transfusiones se requieren solo en raras ocasiones cuando hay hemólisis severa y reticulocitosis inadecuada.
Aunque no hemos visto el frotis de sangre periférica, creo que la información proporcionada es suficiente para concluir que el paciente probablemente tuvo una crisis hemolítica aguda debido a la deficiencia de G6PD, que fue provocada por un factor aún no identificado. Una revisión del frotis de sangre periférica y la medición de los niveles de actividad enzimática pueden ayudar a confirmar el diagnóstico.
DIAGNOSTICO CLÍNICO PROBABLE
ANEMIA HEMOLÍTICA POR DEFICIENCIA DE GLUCOSA-6-FOSFATO DESHIDROGENASA.
DISCUSION PATOLOGICA
un frotis de sangre periférica obtenido el día del ingreso mostró características morfológicas en gran parte normales de las células rojas, con unas pocas células diana. Sin embargo, hubo ocasionalmente posibles células mordidas y células contraídas irregularmente que pueden estar presentes en pacientes con un defecto enzimático de glóbulos rojos, como la deficiencia de G6PD (Figura 1). No se presentaron esquistocitos ni esferocitos. La deficiencia de G6PD se debe considerar en pacientes con anemia hemolítica no esférica aguda y una prueba de Coombs negativa, especialmente aquellos que son hombres de ascendencia africana, mediterránea o asiática.
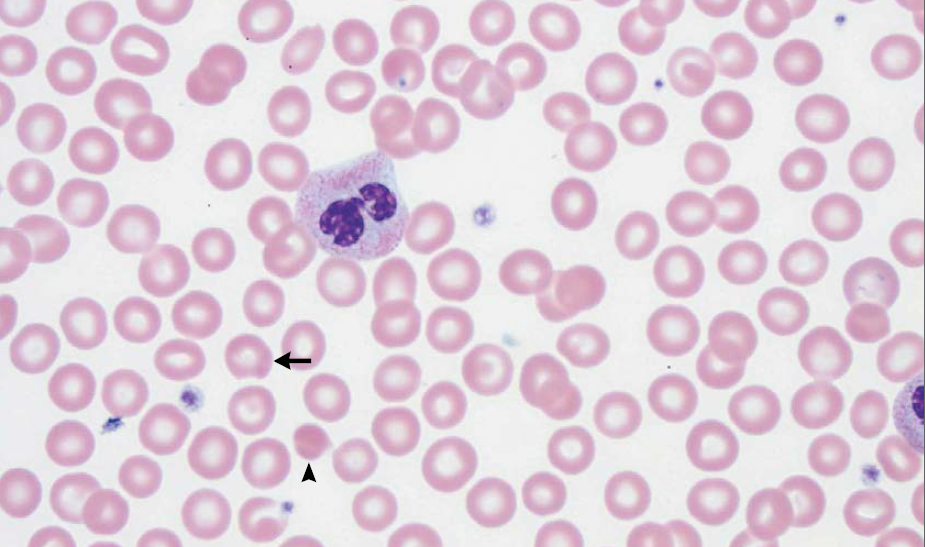
Figura 1 Frotis de sangre periférica (hematoxilina y eosina).
Se obtuvo una muestra de sangre al momento del ingreso. Las características morfológicas de los glóbulos rojos son en su mayoría normales, aunque hay algunas células en diana (target cells), posibles células mordidas (bite cells) (flecha) y células irregularmente contraídas (cabeza de flecha). No se observan esquistocitos ni esferocitos.
La prueba de G6PD se realizó en el hospital el día 4 y reveló un nivel de G6PD de 1.9 U por gramo de hemoglobina (rango de referencia, 8.8 a 13.4). Este nivel es consistente con la deficiencia de G6PD. Dada la hemólisis leve a moderada del paciente y su origen genético, es probable que tuviera la variante A de G6PD, que representa la gran mayoría de los casos de deficiencia de G6PD en personas de ascendencia africana5 (Tabla 3). La variante A− G6PD produce una forma de G6PD que es inestable y, por lo tanto, los niveles de G6PD en glóbulos rojos disminuyen a medida que los glóbulos rojos envejecen. Por lo general, los pacientes con la variante A tienen hemólisis leve a moderada porque solo se ven afectados los glóbulos rojos más viejos. En tales pacientes, el nivel de enzima G6PD es de 10 a 60% de lo normal. Una advertencia importante al realizar pruebas de G6PD es que, en algunas variantes, el nivel de G6PD puede ser normal durante el episodio hemolítico agudo debido a la destrucción de glóbulos rojos más viejos y más deficientes. 6 Por lo tanto, si el nivel de G6PD es normal durante un episodio hemolítico agudo, la prueba de G6PD debe repetirse 1 mes después de que se resuelva la hemólisis. Un problema relacionado es que los pacientes con deficiencia de G6PD y altos recuentos de reticulocitos también pueden tener niveles normales de G6PD debido a los altos niveles de enzimas que se encuentran en los glóbulos rojos jóvenes.

Tabla 3 Variantes genéticas comunes de la glucosa-6-fosfato deshidrogenasa (G6PD).
El paciente permaneció en el hospital durante 4 días. Durante ese tiempo, el hematocrito se mantuvo estable, entre 27% y 31%, y no requirió una transfusión de sangre. Sin ninguna intervención específica, la ictericia se resolvió y los niveles de bilirrubina se normalizaron. Diez días después de su alta hospitalaria, se lo atendió en la clínica ambulatoria, y el hematocrito era del 39,5%, que era su línea de base. Había dejado de tomar ginkgo biloba y se le proporcionó literatura que explicaba qué alimentos y medicamentos debía evitar.
En una visita posterior, el paciente informó de dificultad para respirar y tuvo inquietudes acerca de los sudores nocturnos y la tos. Los resultados de las pruebas de función pulmonar fueron normales. También se realizó una tomografía computarizada de su tórax, abdomen y pelvis, y se observó que tenía un enfisema leve en el lóbulo superior derecho y varios nódulos pulmonares en el lóbulo superior derecho, que medían de 2 a 4 mm de tamaño. Se pensaba que estos representaban granulomas o posiblemente ganglios linfáticos intraparenquimatosos. La evaluación adicional de tuberculosis, infección por hongos y cáncer fue negativa. Las imágenes de seguimiento están programadas para evaluar más a fondo los nódulos.
DUDAS
Dos de los hallazgos en este caso, la esteatosis hepática y el enfisema, pueden estar relacionados con su hábito de fumar, pero ambos también tienen un desequilibrio de oxidación-reducción y una base inflamatoria. ¿Existen informes de daños crónicos debidos a reacciones de oxidación-reducción o estados inflamatorios crónicos en personas con deficiencia de G6PD?
Aunque puede ser tentador atribuir otros aspectos de la presentación del paciente a la deficiencia subyacente de G6PD, estos pacientes suelen ser completamente asintomáticos hasta que sus glóbulos rojos experimentan estrés oxidativo desencadenado por agentes como medicamentos, enfermedades o habas. ingestión. El efecto clínico de la deficiencia de G6PD se limita generalmente a los glóbulos rojos, ya que estas células no pueden producir suficiente NADPH para protegerse del daño oxidativo en ausencia de G6PD.
DIAGNÓSTICO PATOLÓGICO
DEFICIENCIA DE GLUCOSA-6-FOSFATO DESHIDROGENASA.
Traducción de:
“A 29-Year-Old Man with Anemia and Jaundice”
Alberto Puig, M.D., Ph.D., and Anand S. Dighe, M.D., Ph.D.
N Engl J Med 2013; 368: 2502-2509 June 27, 2013DOI: 10.1056/NEJMcpc1302333
References
1
Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician 2004;69: 2599-2606
Web of Science | Medline
.
2
Frank JE. Diagnosis and management of G6PD deficiency. Am Fam Physician 2005;72:1277-1282
Web of Science | Medline
.
3
Yazdi SS, Farajzadeh S, Abadi ACH. Evaluation of G6PD in acne patients. Iran J Dermatol 2010;13:54-56
.
4
Steensma DP, Hoyer JD, Fairbanks VF. Hereditary red blood cell disorders in Middle Eastern patients. Mayo Clin Proc 2001;76:285-293
CrossRef | Medline
.
5
Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008;371:64-74
CrossRef | Web of Science | Medline
.
6
Ringelhahn B. A simple laboratory procedure for the recognition of A− (African type) G-6PD deficiency in acute haemolytic crisis. Clin Chim Acta 1972;36:272-274
CrossRef | Web of Science | Medline