 |
| Hospital "Ángel Pintos" de Azul |
Se internó en nuestro servicio un paciente de 30 años con dificultad en la marcha la cual es cada vez más insegura y parestesias en miembros inferiores de varios meses de evolución. En los últimos 2 meses y sobre todo en los últimos días notó un empeoramiento de la sintomatología motora en ambos miembros inferiores por lo que ayer se decidió consultar. El paciente refiere que suele sentir "como electricidad" en la mitad inferior del cuerpo y que tal sintomatología es fugaz y a veces suele tener relación con movimientos. No refiere trastornos esfinterianos. Pero lo que más le preocupa es la pérdida de fuerzas en miembros inferiores ya que eso le dificulta un trabajo normal (conductor de vehículos). En el examen físico presenta una paraparesia franca, con reflejos osteotendinosos vivos en miembros inferiores y un nivel sensitivo por encima del ombligo, lo que interpretamos como D7, D8 aproximadamente.
Como antecedente de importancia el paciente padece neurofibromatosis tipo 1 (enfermedad de von Recklinghausen), desde temprana edad. La misma es muy florida (imágenes más abajo).
 |
| Manchas café con leche y efélides en NF1. |
 |
| Manchas café con leche, efélides y neurofibromas cutáneos. |
 |
| Manchas café con leche de variados tamaños. Efélides. |
 |
| Neurofibroma cutáneo. |
 |
| Neurofibroma areolar. |
 |
| RMN corte sagital. |

 |
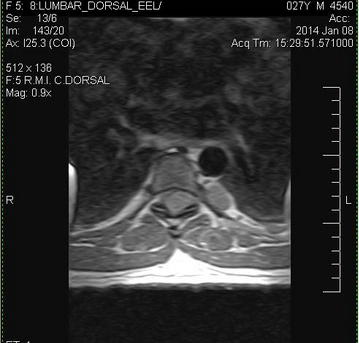
| Un corte axial de RMN donde se ve el neurofibroma atravesandoelagujero de conjunción, invadiendo el canal medular y desplazando a la médula |
 |
| Otro corte donde se ve el neurofibroma |




La neurofibromatosis tipo 1 (NF1) es un trastorno genético con una incidencia de aproximadamente 1 en 2600 a 1 en 3000 individuos. La mitad de los casos son familiares y la otra mitad son el resultado de una mutación esporádica o “de novo”. Los casos de mutación esporádica ocurren en cromosomas derivados del padre y la probabilidad de NF1 tiene relación directa con el aumento de la edad paterna.
NF1 es debido a mutación del gen NF1 localizado en el cromosoma 17q11.2. La neurofibromina, que es la proteína codificada por el gen se expresa en muchos tejidos incluyendo cerebro, riñón, bazo y timo. Pertenece a la familia de las proteínas activadoras de la GTP-asas, que “downregulan” protooncogenes celulares p21-ras codificado por uno de los tres genes RAS, importantes determinantes del crecimiento y la regulación celular. El ras, aciva un número de señales que incluyen el factor de la stem cell (SCF)/c-kit, mTOR y la protein kinasa activada por mitógenos (MAPK). NF1 juega un rol como gen supresor tumoral.
La mutación del gen NF1 produce una pérdida de función de la proteína causando un amplio espectro de hallazgos clínicos entre ellos tumores. El fenotipo de la enfermedad va a estar determinado por la severidad de la mutación, por ejemplo las mutaciones leves (menos de 20 pares de bases), el grado de expresión fenotípica de la enfermedad será leve.
El orden de aparición de las lesiones es: manchas café con leche, efélides o pecas en región axilar y/o inguinal, nódulos de Lisch (hamartomas del iris), y neurofibromas. Las lesiones óseas, si se presentan aparecen durante el primer año después del nacimiento y la hipertensión o la transformación maligna de los tumores puede ocurrir en la adolescencia o en la edad adulta.
Aproximadamente el 25% de los adultos normales tienen manchas café con leche, sin embargo, la presencia de seis o más manchas café con leche son altamente sugestivas de NF1.
Los neurofibromas son los tumores benignos más comunes en NF1 y los gliomas de la vía óptica (GVO) son el tipo de neoplasia intracraneal predominante pero pueden ocurrir otro tipo de neoplasias de SNC o extra SNC.
Los neurofibromas, que son los tumores benignos más frecuentes son tumores de la vaina de los nervios formados por una mezcla de células de Schwan, fibroblastos y células cebadas. Las células de Schwan pueden ser anormales en la NF1 y pueden tener propiedades angiogénicas e invasivas en los neurofibromas plexiformes.
Hay cuatro tipos de neurofibromas:
- Cutáneos
- Subcutáneos
- Plexiformes nodulares
- Plexiformes difusos
Los NEUROFIBROMAS CUTÁNEOS son los más comunes y consisten en tumores blandos pedunculados, generalmente sésiles, similares a los acrocordones. Pueden ser unos pocos hasta varios cientos. Son benignos y no se malignizan nunca. Pueden ser pruriginosos.
Los NEUROFIBROMAS SUBCUTÁNEOS comienzan en la adolescencia o comienzos de la edad adulta, son más firmes de consistencia duro elástica a lo largo de los nervios periféricos. La piel se moviliza por encima de esos nódulos. Pueden ser dolorosos a la palpación.
Los NEUROFIBROMAS PLEXIFORMES NODULARES desarrollan debajo de la dermis en los tejidos profundos aparecen como como grupos de neurofibromas a lo largo de las raíces de los nervios, son similares a los neurofibromas subcutáneos pero no se palpan. A diferencia de los plexiformes difusos los nodulares no comprometen ni invaden otros tejidos tales como el músculo, vasos o partes blandas. Pueden comprometer la columna y producir déficits neurológicos.
Los NEUROFIBROMAS PLEXIFORMES DIFUSOS, son considerados como lesiones congénitas, aunque no aparecen en los infantes. Hay hiperpigmentación de la piel en la zona donde más tarde desarrollará la lesión con los años. La mayor diferencia entre este tipo de neurofibromas y otros es que estos invaden los tejidos que lo rodean haciendo más dificultosa la resección completa
Los tumores malignos de las vainas de los nervios antes llamados neurofibrosarcomas usualmente asientan o se originan en neurofibromas plexiformes preexistentes que se someten a transformación maligna.
Todos los neurofibromas plexiformes, tanto los nodulares como los difusos comprometen la mayoría de las veces varios fascículos nerviosos, con un crecimiento serpiginoso y vascularización significativa. Esas lesiones pueden ser un desafío en el tratamiento quirúrgico y el manejo del dolor, especialmente cuando crecen cercanos a la columna resultando en erosión vertebral o compresión de la médula espinal.
Nuestro paciente padece un neurofibroma plexiforme nodular de la raíz nerviosa D6 que se proyecta al canal medular a través del agujero de conjunción, desplazando la médulaespinal y causando paraparesia. Por tal motivo está indicada la cirugía de la lesión con liberación del canal.