Paciente masculino 17 años
MOTIVO DE INTERNACIÓN: pérdida de conocimiento, cefalea, fiebre.
ENFERMEDAD ACTUAL: comienza hace 15 días con cefalea frontotemporal izquierda de una intensidad en los momentos de mayor dolor de 8 en una escala de 1 a 10 en la que 10 es dolor intolerable. Comienza por ese motivo a automedicarse con paracetamol e ibuprofeno a demanda. El paciente no presenta cefalea habitualmente siendo sano según el interrogatorio. Casi simultáneamente con la cefalea comienza a presentar resfrío con secreciones nasales purulentas sin sangre y no demasiado abundantes. Hace 7 días consulta al médico dado la persistencia del cuadro doloroso diagnosticándose sinusitis y prescribiéndose ATB que el paciente no comienza a tomar porque en la farmacia solicitan la medicación a una droguería por no contar con la misma en ese momento (vive en un poblado rural pequeño). El paciente refiere en las últimas 48 horas, además de la persistencia de la cefalea, episodios de escalofríos seguidos de probable alza térmica nunca documentada.
Hace 72 horas presentó pérdida de conocimiento estando acostado mirando televisión siendo trasladado por un servicio de emergencias de una localidad vecina al hospital local y posteriormente derivado a nuestro hospital. El paciente no sabe cuánto tiempo estuvo inconsciente pero fueron no menos de 20 minutos presentando amnesia completa del episodio. El mismo estuvo presenciado por un familiar quien refiere actividad motora involuntaria fugaz sugestiva de cuadro comicial. Después del episodio presentó sangrado de lengua por lesión traumática durante la actividad convulsiva. No existen otras lesiones traumáticas como consecuencia de la pérdida de conocimiento ya que el paciente estaba acostado. Al llegar a nuestro hospital se lo somete inmediatamente a estudios de imágenes (TC,RMN).
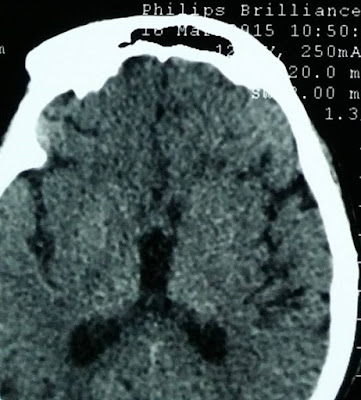
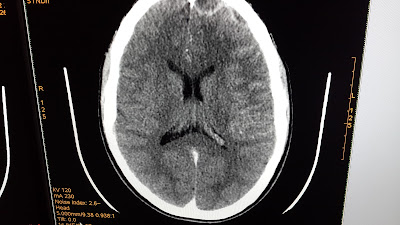
TC de cráneo donde se observan dos imágenes en situación extraaxial (epidural), en la región frontal anterior izquierda, que refuerza en la periferia con la administración d contraste sugestiva de empiema
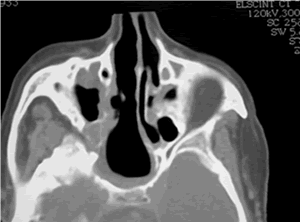
Rx simples de cráneo localizadas en región frontal donde se observa ocupación de los senos frontales sin la aireación fisiológica o normal. Mucocele frontal
Solución de continuidad de la estructura ósea en relación con el seno frontal. Mucocele frontal
En RMN se ven las dos imágenes vistas en TC hipointensas en T1,hiperintensas en T2.Se aprecia mucocele en seno frontal en las últimas dos imágenes
Reconstrucción 3D en TC que muestra una zona de solución de continuidad a nivel del hueso frontal con el espacio epidural.
ANTECEDENTES PATOLÓGICOS Y FAMILIARES: NO REFIERE
EXAMEN FÍSICO:
Al ingreso a nuestro hospital el paciente estaba lúcido, ubicado en tiempo y espacio pero con letargia, somnolencia, y con amnesia del episodio anterior, febril 38,2 °C, y se quejaba de cefalea.
TA 120/80 mmHg, frecuencia cardíaca 82 por minuto regular. Frecuencia respiratoria 18 por minuto.
Pupilas iguales reactivas. No hay foco neurológico. No hay síndrome meníngeo.
Abdomen: s/p
Tórax: s/p
Cardiovascular: s/p
LABORATORIO:
GR: 4170000. GB 12300 ( segmentados 84%,linfocitos 10,monocitos6).
Hepatograma: s/p
Función renal normal. Coagulograma normal. Ionograma normal (Na 142, K 4,3, Cl 109).
VIH negativo
El paciente fue intervenido quirúrgicamente de un mucocele de seno frontal y drenado un absceso epidural evacuándose pus franco que mostró abundantes cocos grampositivos en el examen directo pero que no desarrollaron en los cultivos. La interpretación fue que probablemente el agente etiológico puede haber sido un anaerobio que es en sí mismo de difícil desarrollo in vitro sumado a la presión antibiótica de 15 días al momento de la cirugía.
El paciente tuvo una muy buena evolución post operatoria recuperándose satisfactoriamente. Continuará con tratamiento antibiótico por el término de 8 semanas.
CONCLUSIONES DEL CASO
Este paciente de 17 años con antecedentes de sinusitis a repetición presentó una complicación de un mucocele de seno frontal hasta ese momento desconocido que fueron dos abscesos epidurales intracraneales por comunicación del mucocele con la cavidad intracraneal en la fosa cerebral anterior. Es sabido que los mucoceles frontales (por otro lado la localización más frecuente de los mucoceles de senos paranasales), hacen su crecimiento predominantemente hacia la órbita muchas veces desplazando al globo ocular hacia adelante y hacia abajo. Sin embargo está descripta la complicación de absceso intracraneal epidural como presentó este paciente, así como también el compromiso subdural, meningitis etc como veremos más abajo en el desarrollo de mucoceles de senos paranasales. El paciente fue tratado médicamente con ATB y posteriormente sometido a cirugía del mucocele y drenaje del absceso epidural con excelente evolución.
ABSCESO EPIDURAL INTRACRANEAL
Los abscesos epidurales intracraneales (AEI) son menos comunes que los abscesos epidurales espinales (AEE), y menos aguda su evolución. Sin embargo, como Los AEE , son infecciones que requieren tratamiento óptimo y rápido para prevenir complicaciones.
ANATOMÍA
La duramadre intracraneal forma el revestimiento interior del cráneo y está directamente adherida al hueso. Por lo tanto, en circunstancias normales, no hay espacio epidural real. El potencial espacio epidural puede ser abierto por la presión de expansión de tumores, sangre, masas inflamatorias, o pus. Esto requiere que la duramadre firmemente adherida se despegue del hueso; Como resultado de ello, el AEI tiende a ser de crecimiento lento, redondeado, y bien localizado.
PATOGENIA Y PATOLOGÍA
Los organismos generalmente se propagan al espacio extradural potencial por extensión directa desde un foco contiguo de infección o por inoculación durante un trauma o neurocirugía. Después de llegar a este sitio, las bacterias causan inflamación y la formación de pus o tejido de granulación, que diseca gradualmente la dura resistente y adherente separándola de la tabla interna del cráneo.
AEI generalmente consisten en una lesión localizada con una central de de pus rodeada por un muro de reacción inflamatoria (que puede calcificarse). Rara vez se extienden hacia abajo (caudal) desde el interior del cráneo hacia el canal espinal debido a que la duramadre está muy estrechamente unida alrededor del agujero occipital. Estos resultados son diferentes de las AEE en el que el tejido de granulación en lugar de pus es un hallazgo frecuente y l propagación longitudinal de la infección es prácticamente la regla.
EPIDEMIOLOGÍA AEI es la tercera infección intracraneal focal más común, después de absceso cerebral y empiema subdural. En el pasado, la mayoría de los casos se asociaron con sinusitis, otitis, o mastoiditis. Hoy en día, muchos casos surgen como una complicación de los procedimientos neuroquirúrgicos.
MICROBIOLOGÍA Y PUERTAS DE ENTRADA
La puerta de entrada usual de entrada es desde el exterior. Si la infección inicial está en los senos paranasales o los oídos, los organismos es probable que sean los estreptococos microaerofílicos o anaeróbicos y / u otros anaerobios tales como especies de Propionibacterium y Peptostreptococcus. Unos pocos casos se deben a bacilos gramnegativos aeróbicos u hongos.
Si la infección sigue a neurocirugía, los más probables organismos son estafilococos, especialmente S. aureus, y bacterias gram-negativas. La infección también puede propagarse hacia el interior desde la osteomielitis del cráneo o ser introducido por sondas de monitoreo fetal aplicadas al cráneo durante el parto.
MANIFESTACIONES CLÍNICAS
Signos y síntomas son el resultado de la infección y la masa intracraneal de lento crecimiento y expansión. Esto último con el tiempo puede causar aumento de la presión intracraneal, edema de papila, y, en algunos casos, signos neurológicos focales. Fiebre, dolor de cabeza, letargo, náuseas y vómitos son comunes. El AEI en el contexto de infecciones de los senos pueden causar drenaje purulento de la nariz o el oído.
Debido a una AEI se presenta generalmente como complicación de otro proceso, la infección primaria puede convertirse en el foco de atención y retrasar el diagnóstico de la extensión intracraneal. A modo de ejemplo, la inflamación local y dolor pueden ser atribuidos a un trauma reciente, neurocirugía, o sinusitis, cuando en realidad se superpone a una AEI.
DIAGNÓSTICO
La clave del diagnóstico es considerar esta condición rara, a continuación, llevar a cabo un examen físico seguido de la imagen apropiada. La resonancia magnética (RM) por lo general proporciona más información que la tomografía computarizada (TC). La aspiración con aguja guiada por TC o drenaje abierto pueden proporcionar material para la tinción y cultivo para bacterias, micobacterias y hongos.
Un AEI puede imitar cualquier lesión de masa intracraneal, incluyendo primaria del parénquima y los tumores metastásicos, meningiomas, hematomas, abscesos cerebrales, o empiemas subdurales. Meningitis crónica, meningitis tuberculosa, o arteritis craneal también deben ser considerados.
Manejo
El tratamiento exitoso de un AEI requiere una combinación de un procedimiento de drenaje y tratamiento antibiótico. Drenaje de Neurocirugía se requiere generalmente a través de los agujeros de trépano o una craneotomía. Si la duramadre está macerada o rota, un injerto fascial se puede aplicar.
El tratamiento antibiótico empírico debe elegirse con base en el origen probable de la infección. Si la infección contigua de los senos paranasales, oído, o mastoides es la fuente, un régimen que sea activo contra estreptococos, especies de Haemophilus, y anaerobios deben ser elegidos.
El tratamiento antibiótico más específico se debe dar después de que los resultados de las tinciones y cultivos indiquen el organismo (s). Con la administración de antibióticos y quirúrgico adecuado, el pronóstico de la AEI es bueno, con una mortalidad atribuible por debajo del 10 por ciento.
TERAPIA ANTIMICROBIANA
TERAPIA EMPÍRICA
Un régimen empírico con antibióticos activos contra estafilococos, estreptococos y bacilos gramnegativos debe ser elegido. Los antibióticos deben dirigirse contra el patógeno conocido si la cultura y / o tinción de Gram del aspirado es positivo. Si un aspirado no se puede obtener, el régimen empírico debe continuar.
Regímenes parenterales empíricos apropiados incluyen:
- La vancomicina (30 a 60 mg / kg por vía intravenosa [IV] diaria en dos dosis iguales ajustados para la función renal) para la cobertura empírica de MRSA y para su uso en pacientes que son alérgicos a la penicilina. Si las pruebas de sensibilidad revela sensible a meticilina S. aureus (MSSA), la vancomicina debe ser reemplazado con nafcilina (2 g IV cada cuatro horas) O oxacilina (2 g IV cada cuatro horas) en pacientes sin alergia a estos agentes, ya que tienen mejor penetración al sistema nervioso central (SNC).
- MÁS
- Metronidazol (500 mg IV o por vía oral cada ocho horas)
- MÁS
- ● Cefotaxima (2 g IV cada seis horas), o ceftriaxona (2 g IV cada 12 horas), o ceftazidima (2 g IV cada 8 horas). La ceftazidima es preferible cuando Pseudomonas aeruginosa se considera un posible o probable patógeno.
DURACIÓN
La duración habitual del tratamiento es de seis a ocho semanas o hasta que la resolución de la SEA en la RM. La primera resonancia magnética de seguimiento se obtiene en aproximadamente cuatro a seis semanas si el paciente está mejorando o en cualquier momento si se produce un deterioro clínico.
PRONÓSTICO
Alrededor del 5 por ciento de los pacientes con AEI pueden morir debido a sepsis no controlada u otras complicaciones.
MUCOCELES DE SENOS PARANASALES.
En 1818 Langenbeck describió por primera vez las características clínicas, Rollet, en 1896, fue el introductor del término mucocele y la primera descripción histológica la hizo Onodi en 1901.
Se trata de un proceso patológico crónico pseudotumoral debido a la acumulación y retención de material mucoide dentro de un seno por obstrucción continúa o periódica del ostium de drenaje con la consecuente dilatación del mismo.
La mayoría de los mucoceles ocurren entre la cuarta y séptima década de la vida, aunque en la literatura se describen casos en edades más tempranas. Es raro la aparición de mucoceles frontales antes de los 10 años, debido a que en esa edad no se ha desarrollado por completo dicho seno paranasal.
Es raro que se manifiesten de forma bilateral, en las distintas series no hay diferencias significativas en cuanto al sexo.
Todos los senos paranasales pueden desarrollar un mucocele, aunque los más frecuentes son los de ubicación frontoetmoidal, siendo excepcionales aunque no infrecuentes, los de origen esfenoidal y maxilar.
Independientemente de su localización, todos tienen dos fases: la primera paucisintomática o de latencia clínica. La segunda fase de exteriorización o complicación donde el mucocele se expande fuera de los límites del seno involucrado.
El tratamiento siempre debe ser quirúrgico, dependiendo la vía de acceso de la localización y el compromiso de estructuras vecinas, con la colaboración en muchos casos de neurocirujanos u oftalmólogos entrenados en dicha patología.
El problema más grave del tratamiento es la recidiva.
DEFINICION
Son formaciones pseudoquísticas expansivas de los senos paranasales con retención de secreción mucosa. Su pared está constituida por la mucosa más o menos modificada de la cavidad sinusal y su contenido es un líquido aséptico, en general, de consistencia espesa. Dentro del seno afectado se produce un aumento de presión que genera la dilatación progresiva de la pared del mismo lo cual determina el adelgazamiento de su pared con fenómenos de remodelación ósea.
INCIDENCIA
En todas las series analizadas la repartición por sexo es igual y la edad de aparición suele ser entre los cuarenta y cincuenta años. No hay diferencias étnicas descriptas.
El 80% se localizan a nivel frontal y etmoidal (siendo el primero más frecuente), aproximadamente un 10% se ubican en el seno esfenoidal y maxilar.
Más frecuentes unilaterales.
ANATOMIA PATOLOGICA
Constituídos por tejido conectivo denso frecuentemente hialinizado. En su interior esta tapizado por epitelio pseudo estratificado o columnar bajo que ocasionalmente contiene células caliciformes, sin cilios con hipertrofia de estructuras glandulares. Contenido líquido, espeso, filante, amarillento o grisáceo y estéril, que contiene gotas de grasa, células epiteliales y mucina.
Una complicación posible es la formación de un mucopiocele. Los estudios microbiológicos actuales demuestran la predominancia de organismos que forman parte de la flora orofaríngea (Peptostreptococcus sp, Provotella sp, Fusobacterium sp) aunque en la mayoría de los casos se aislan conjuntamente aerobios y anaerobios.
La frecuencia de anaerobios en el mucopiocele es debida a cambios en el pH y a la disminución de la concentración de oxígeno en la cavidad sinusal secundaria a la obstrucción del ostium y a los procesos infecciosos de la mucosa sinusal.
ETIOLOGIA
Varias teorías han sido postuladas para explicar el origen de esta patología: congénita, infecciosa, traumática, inflamatoria, pero aún así, en muchos casos permanece desconocido. En la actualidad, se cree que es secundaria a la asociación de dos fenómenos:
1) obstrucción del ostium de drenaje
2) inflamación.
Sin precisarse en todos los casos cuál de los dos es el primer evento en producirse. El bloqueo del ostium produce en la mayoría de los casos un acúmulo de material mucoso intrasinusal, pero solo una pequeña proporción progresa a la formación de un mucocele.
Existen varios factores que predisponen a la formación de los mucoceles, pudiendo dividirse éstos en dos grupos: extrínsecos e intrínsecos. Dentro de los extrínsecos: traumatismos, antecedentes quirúrgicos previos, poliposis nasal, tumores benignos, tumores malignos y alteraciones anatómicas.
Intrínsecos: cualquier situación que aumente la viscosidad o éctasis de las secreciones nasosinusales: Fibrosis Quística, enfermedad alérgica nasosinual, sinusitis crónica, infecciones recidivantes, hipersecreción mucosa.
Se han relacionado también: la obstrucción del conducto de las glándulas salivales accesorias localizadas dentro de la cubierta del seno paranasal y niveles elevados de prostaglandina E2 como agentes causales. Existen estudios histológicos que han confirmado la presencia de fenómenos de remodelación del hueso que se encuentra en contacto con el mucocele debido a factores de reabsorción ósea (PGE2, colagenasa, ILK1 y TNF) liberados por la mucosa cuando esta se ve sometida a procesos de tipo inflamatorio. Esta remodelación es compensada por proliferación osteoblástica.
PRESENTACION CLINICA
Independientemente de su localización, todos, tienen dos fases:
1) Fase inicial: que se constituye dentro de la cavidad sinusal. Es el período de latencia clínica o paucisintomático.
2) Fase de exteriorización o complicación: el mucocele se expande fuera de los límites del seno. Se erosionan las paredes óseas poniéndose en contacto su pared con las estructuras vecinas. La clínica dependerá del asiento y sitio de partida.
Según su localización los mucoceles se dividen fundamentalmente en tres tipos, cada uno de ellos con implicancias clínicas y terapéuticas diferentes.
I. Los mucoceles originados en el sistema frontoetmoidal o anterior: son los más frecuentes. En el período inicial se caracterizan por ser asintomáticos o con clínica banal. El período de exteriorización lo dominan los signos oftalmológicos, quedando los rinológicos en segundo plano.
Consulta por cefalea frontal y opresión cefálica. Epífora. Exoftalmos: desplazamiento del globo ocular hacia abajo y afuera (Fig.1). Diplopía y atrofia del Nervio Óptico.
Figura 1: Exoftalmos: desplazamiento del globo ocular hacia abajo y afuera. Mucocele frontoetmoidal anterior.
Los mucoceles del seno frontal pueden erosionar la tabla anterior y se exterioriza como una tumoración en el ángulo supero interno de la órbita, blando e indoloro y renitente: Tumor algodonoso de Pott (Fig.2) que desplaza el globo ocular y puede fistulizar a piel. Si erosiona la pared posterior frontal: absceso epidural, subdural, cerebral y meningitis.
Figura 2: Tumor algodonoso de Pott en un mucocele frontal.
Los de localización etmoidal, destruyen la lámina papirácea (cara medial de la órbita) y el globo se desplaza hacia fuera o abajo. Compresión de la hendidura esfenoidal, parálisis ocular y disminución de la sensibilidad frontal.
La sintomatología rinosinusal suele asociar: insuficiencia ventilatoria nasal unilateral e hiposmia.
II. Los mucoceles originados en el sistema posterior o etmoidoesfenoidal: son menos frecuentes. Se caracterizan por una larga fase de latencia. El desarrollo del etmoides posterior depende fundamentalmente de la Celda de Onodi y el mucocele esfenoidal, más frecuente, aparece muchas veces bilateral por la fragilidad del septum intersinusal pudiendo alcanzar un volumen de 500 ml (Fig. 3)
Figura 3: Mucocele esfenoidal
Se extienden lateralmente hacia la región del ápex orbitario con el canal óptico hacia arriba y adelante, la Hendidura Esfenoidal con los nervios oculomotores más abajo y el seno cavernoso detrás. Los pares II, III y IV son los más vulnerables.
Pueden evolucionar hacia delante donde la región nasoetmoidal ofrece poca resistencia, pero la tolerancia es buena, hacia arriba: hipófisis o quiasma (tumor expansivo) o hacia abajo: cavum.
Las manifestaciones clínicas son de dos tipos fundamentalmente: Algias del vértex craneano, periorbitarias, retrooculares y cefalea occipital: el síntoma más precoz y frecuente. Los síntomas oftalmológicos, que suelen ser más tardíos: exfoftalmos, diplopía, parálisis oculomotoras sobre todo del III par, compresión y atrofia de quiasma y Nervio Óptico. Rinorrea cerebroespinal.
La clínica rinológica es menos llamativa: anosmia, insuficiencia ventilatoria nasal. Tardíamente se puede observar hipopitituarismo y complicaciones endocraneales.
III. Los mucoceles del seno maxilar: son excepcionales. Representan menos del 10% de los casos ya que son cavidades amplias y bien ventiladas. En el 50% de los casos hay antecedentes de cirugías previas (Caldwell- Luc) que han dejado como secuelas bridas cicatrizales.
Presentan pobre sintomatología cuando se localizan dentro del seno: rinorrea seromucosa, cefalea frontal, obstrucción nasal y sensación de presión o dolor en la mejilla.
Cuando exceden los límites del seno: tumoración de tejido blando indolora,
exoftalmos con desplazamiento del globo ocular hacia arriba, destrucción del piso orbitario, diplopía, paresia del III par craneal y del Nervio Óptico. Si se colapsa espontáneamente luego de adelgazar el piso orbitario: enoftalmos y diplopía. Además se puede extender hacia la fosa pterigomaxilar, región endobucal, endonasal y puede comprimir el Nervio Infraorbitario manifestándose como neuralgia.
IV. Otras localizaciones: cornete inferior, cornete medio y fosa pterigomaxilar. El de cornete medio es el más frecuente y predomina en pacientes que presentan un cornete bulloso (incidencia de 4-14% de neumatización del cornete medio en la población general).
Se presenta como un tumor endonasal que produce obstrucción nasal crónica unilateral o bilateral con desviación septal y compresión de estructuras vecinas cuando tienen mucho tiempo de evolución. A la inspección endoscópica se presenta como una masa redondeada cubierta por mucosa nasal normal de consistencia firme. En la Tomografía masa expansiva con centro hipodenso que no refuerza con el contraste cuyo diagnóstico diferencial es con un tumor que lo diferencia por la delgada pared del cornete medio (Fig. 4).
Figura 4. Mucocele del cornete medio
El tratamiento es endoscópico con extirpación del tumor, meatotomía media, drenaje del seno maxilar en caso de que esté ocupado y apertura de las celdillas etmoidales anteriores.
DIAGNOSTICO
La prueba diagnóstica de elección es la Tomografía Computada complementada con la Resonancia Magnética Nuclear.
La TAC permite visualizar el contenido iso o hipodenso homogéneo con relación al parénquima cerebral que no capta contraste, solo el piocele se tiñe periféricamente (Fig. 5).
Figura 5. Mucopiocele frontal izquierdo con realce periférico en la TC con contraste.
Permite estudiar las diferentes reacciones de las paredes óseas: lisas, netas, delgadas de forma redondeada u oval (Fig. 6).
Figura 6.
Otros casos son irregulares y poco visibles. Puede formarse una banda gruesa y densa de osteocondensación por la reacción osteoblástica. Imágenes de invasión y osteólísis que plantean el diagnóstico diferencial con neoplasias las cuáles muestran una imagen heterogénea que realza con el contraste administrado.
La RMN evalúa las relaciones del mucocele con las partes blandas cerebrales, orbitarias y vasculares. Aprecia la topografía y morfología del mucocele. El contenido presenta dos patrones diferentes de señal de acuerdo con su estado de hidratación y su contenido proteico y mineral.
Un patrón es hiperintenso en T1 debido a un alto contenido proteico, e hiperintenso en T2 por su alto contenido acuoso. Estos mucoceles en Tomografía se ven moderadamente densos. (Fig. 7, 8 y 9)
Fig. 7. RM en T1: hipointenso en T1 por su alto contenido proteico
Fig. 8. T1 luego de inyectar Gadolinio: mucosa teñida hipointensa
Fig.9. RM en T2: lesión hiperintensa por su alto contenido en agua.
El otro patrón es hipointenso en T1 y T2. Corresponde a mucoceles envejecidos, deshidratados, con detritus seco y depósitos de sales cálcicas, de potasio, de zinc, hierro, magnesio y manganeso. Algunos autores han detectado la presencia de Aspergillus. La mayoría son hiperdensos en tomografía, debido a la presencia de calcio distribuido en forma difusa. La RMN puede ayudar a diferenciar entre un tumor sólido del contenido fluido o mucoso que puede mostrar un seno ocupado con un proceso inflamatorio: por ejemplo con técnicas de supresión de la grasa.
Las Rx simples muestran generalmente la opacidad sinusal, cuyo contorno óseo suele estar alterado con signos de osteólisis u osteocondensación. Cuando son expansivos se observa el desplazamiento de la pared ósea.
Algunos mucoceles frontales no opacifican el seno (4), tienen por el contrario una llamativa hipertransparencia que sumada a la erosión y la expansión las paredes dan una imagen en “globo aéreo” (Fig. 10).
Figura 10. Algunos mucoceles frontales no opacifican el seno sino que por el contrario tienen una llamativa hipertransparencia que da la imagen en "globo aéreo"(asterisco)
El mucocele antral típico opacifica por completo el seno maxilar, expande la cavidad sinusal, las paredes óseas evidencian osteocondensación o erosión de la misma. En la tomografía se observa abombamiento de la pared medial del seno o erosión de la apófisis unciforme.
En el caso del mucocele del seno maxilar la endoscopia preoperatoria en muy valiosa en el diagnóstico temprano del mismo. La tríada de: abombamiento de la pared medial del seno, prolapso de la mucosa del meato medio con obstrucción del mismo y drenaje purulento a través del mismo es altamente sugestivo de mucocele antral.
TRATAMIENTO
Preventivo: consiste en el control de las infecciones y de las poliposis desde el punto de vista médico y el respeto de las estructuras fundamentales en las intervenciones quirúrgicas, hecho que se consigue más fácilmente con la cirugía endoscópica funcional.
Curativo: siempre debe ser quirúrgico. Se debe conocer muy bien su extensión y la afectación de vecindad. A veces se requerirá la colaboración de neurocirujanos y oftalmólogos.
MUCOCELES POSTERIORES
Las lesiones oculares dominan el pronóstico y pueden conducir a una cirugía de urgencia.
La mayoría de los autores, consultados en las distintas bibliografías, están de acuerdo que el tratamiento de los posteriores, sea cual sea su extensión, es la Marsupialización Endoscópica ( apertura amplia del seno con remoción de la pared anterior e inferior del mucocele y favorecer la amplia comunicación del seno afectado con las coanas y fosas nasales.
Antes de proceder a la apertura del seno esfenoidal. En primer lugar se comprobará con la tomografía la relación del nervio óptico y arteria carótida interna con las paredes sinusales y la distancia que existe entre el vestíbulo nasal y la pared anterior del seno.
El abordaje se puede realizar por tres vías distintas:
1. Vía transseptal: si previamente se ha realizado septumplastia. Vía segura porque realiza la entrada en la porción más medial y declive del seno, pero presenta dificultades técnicas debido al sangrado de la mucosa septal y la dureza de la pared anterior del seno.
2. Vía transnasal: a través del receso esfenoetmoidal. Punto de referencia es el reborde superior de la coana se asciende junto al tabique unos 10 cm. por encima. En la pared lateral se tiene la cola del cornete medio y superior. En ocasiones se visualiza el ostium natural que se amplía en sentido descendente con una pinza de Kerrison. Nunca se debe ampliar en sentido ascendente ya que a 1 cm. de distancia se ubica la lámina cribosa. En la mayoría de los casos no se localiza el ostium natural y se realiza la entrada al seno en la pared anteroinferior de seno a unos 10 mm del reborde superior de la coana. Para lograr una amplia visión, Wigand propone resecar sistemáticamente la cola del cornete medio y la septoplastia. Esta vía permite realizar una esfenoidotomía aislada en caso de una patología esfenoidal circunscripta
3. Vía transetmoidal: es la más difícil ya que desde el etmoides posterior se carece de puntos de referencia. Se puede tomar como referencia el reborde superior de la coana y realizar la apertura en la parte más declive y medial de la pared anterior del seno para evitar traumatizar su pared lateral en la que se destacan los rebordes del nervio óptico y de la arteria carótida interna.
Sea cual sea la vía de acceso, se amplía la apertura del seno para permitir el correcto drenaje y aireación del mismo y se extirpan las lesiones con sumo cuidado y sin brusquedad para favorecer la amplia marsupialización hacia la fosa nasal. TAC. Postoperatoria que evidencia la marsupialización. (Fig. 11)
Figura 11.
MUCOCELES ANTERIORES
Hay dos tendencias dispares: una, la técnica conservadora, marsupializando el mucocele y repermeabilizando el ostium frontal por vía externa o endoscópica. La otra, técnica radical con exéresis completa del mucocele y exclusión del seno afectado sobre todo si hay extensión orbitaria o endocraneal.
Como primer paso es esencial la delimitación por tomografía de la anatomía rinosinusal.
A continuación se detallará las distintas vías de acceso externa y endonasal, con la descripción de las técnicas básicas para el seno frontal donde la patología es más frecuente.
Vía de acceso al seno frontal externa:
— Vía frontosuperciliar: otorga un acceso amplio al seno frontal, permite tratar las lesiones endosinusales, y si es necesario un buen acceso al conducto nasofrontal. Existen numerosas variantes técnicas dependiendo que se realice o no una ventana osteoplástica (ósea u osteoperióstica).
Se realiza una incisión siguiendo el borde inferior de la ceja, que nunca se debe rasurar. Comienza sobre el hueso propio frente al canto interno y se extiende hasta el límite externo del seno frontal delimitado radiológicamente. Afecta piel, planos musculares y periostio. Se realiza un raspado subperióstico para exponer la cara anterior del seno, hacia abajo y adentro el límite es la polea del músculo oblicuo mayor. La abertura sinusal se puede realizar por trepanación simple o confeccionar una ventana ósea a expensas de la pared anterior del seno. La primera se realiza con una fresa que genera una pérdida de sustancia ósea y la visión directa es reducida por lo tanto no apta para el tratamiento del mucocele. La segunda técnica consiste en confeccionar una ventana ósea mediante punteado con fresa cuyas dimensiones dependen del tamaño del seno realizando luego la osteosíntesis con hilos de acero. No se deja drenaje sinusal transcutáneo. El principio de la ventana permite una cirugía sinusal completa, conservando al máximo la mucosa sana, repermeabilización del conducto nasofrontal y no crea defecto óseo. También se puede optar por una cirugía de exclusión del seno o se pueden realizar colgajos osteoperiósticos para preservar la vascularización ósea.
A veces la incisión se puede realizar más alta y bilateral para acceder a ambos senos frontales.
— Vía coronal de Unterberger: vía de acceso amplia y directa a los dos senos frontales, que libera toda la cara anterior del cráneo y permite descender hasta la punta de la nariz.
Incisión arciforme cinco cm. por detrás de la implantación del cuero cabelludo, se incide hasta periostio y se confecciona la ventana ósea que posteriormente se cierra como en la técnica anterior. Complicaciones: anestesia frontal, parálisis frontal y problemas estéticos con la cicatriz en calvos o con predisposición familiar o personal de calvicie.
Vía de acceso endonasal:
Permite acceder al conducto nasofrontal y al piso del seno frontal, permitiendo el tratamiento fisiológico de la mucosa del seno frontal. Además es un soporte valioso en los abordajes externos para el tratamiento del etmoides y conducto nasofrontal. La cirugía endoscópica funcional de los senos paranasales (FESS) ha sido aceptada como el mejor procedimiento para el tratamiento quirúrgico de la sinusitis crónica. Se propone extirpar sólo el tejido necesario para aliviar la obstrucción y conservar la mucosa de la vía de drenaje para evitar la reestenosis. La sinusotomía frontal endoscópica, en los centros especializados, ha sobrepasado en su resultado a los métodos externos con menor morbilidad, convalecencia menor y no deja cicatrices en la piel. (16)
El seno frontal es el más complejo de los cuatro senos paranasales para el tratamiento endoscópico. Su localización anterosuperior y la complejo anatomía del relativamente estrecho conducto nasofrontal dificulta la visualización y predispone a la estenosis.
Durante el examen endoscópico del meato medio, encontramos entre la cabeza del cornete medio y la apófisis unciforme una estructura redondeada que contiene una celda: bulla etmoidal. Las prolongaciones superiores del cornete medio, apófisis unciforme y la bulla se reúnen en forma de estrella: “encrucijada bullar”. Entre ellas delimitan tres canales:
— Anterior: delimitado por la apófisis unciforme y el cornete medio: en su porción superior se localiza la desembocadura del conducto nasofrontal.
— Lateral: entre la apófisis unciforme y la bulla, en su porción inferior se encuentra el ostium del seno maxilar.
— Medial o Retrobullar: entre la bulla y el cornete medio: ostium de las celdillas etmoidales anteriores.
Por detrás de la bulla, los canales lateral y medial se unen formando un canal único que desemboca en la coana.
Es fundamental conocer profundamente la anatomía endoscópica ya que la dificultad de acceso al conducto nasofrontal varía con la extensión de la patología etmoidal y con la neumatización del receso frontal.
ACTITUD ANTE LA FUNCIÓN SINUSAL
Ya mencionamos anteriormente, que existen dos actitudes opuestas en el tratamiento de los mucoceles anteriores: la preservación o la exclusión del mismo. El elemento decisivo es el estado del conducto nasofrontal.
La cirugía funcional necesita un drenaje y una aireación eficaz a través de un conducto de tamaño suficiente favoreciendo el drenaje sinusal y extirpando las lesiones. Es preciso distinguir dos situaciones: conducto permeable, sin lesiones que estén en contacto con su ostium con mucosa indemne.
En este caso se debe ser conservador sin traumatizar la mucosa y preservar como mínimo un collar de mucosa sana alrededor del orificio superior del conducto.
La segunda situación nos encontramos con un conducto no permeable obstruido por la patología, aquí se debe repermeabilizar por vía externa o endoscópica. Schaefer utiliza sistemáticamente un tubo de silicona de 4 mm de diámetro cuando el orificio del canal es menor a 6 o 4 mm y lo mantiene por seis semanas. Har-El utiliza una sonda de intubación nasotraqueal número 3 a 4,5. Yamasoba usa un tubo en T de Montgomery de 8 a 10 mm en caso de recidiva luego de la primera operación y lo mantiene por 6 meses. Amble y Neel utilizan una sonda de silicona flexible que no genera necrosis ni osteogénesis como en el caso de las sonda rígidas con la consiguiente recidiva. También se puede realizar el drenaje utilizando el conducto nasofrontal contralateral que debe ser permeable y se suprime el tabique intersinusal, obviamente que se debe tratar de una patología etmoidofrontal unilateral.
La exclusión exige la supresión total del conducto nasofrontal. Esta técnica conserva su papel en la patología inflamatoria en el caso en el que el seno operado varias veces con métodos “conservadores” es el asiento de recidivas. Consiste en acceder a los senos frontales por vía coronal, confeccionar una ventana ósea a su nivel, resecar la integridad de la mucosa de los dos senos frontales y obturar el conducto nasofrontal por invaginación de la mucosa y colocar un tope óseo. Se rellenan ambos senos frontales con hidoxiapatita, matriz colágena, esponja de silicona, músculo, hueso esponjoso o grasa autóloga que es el único material inocuo. Se cierra la ventana y se dejan dos drenajes. Conlleva tres tiempos quirúrgicos:
1. Ablación total de la mucosa del seno: es esencial quitarla en su totalidad con coagulación, fresado, o raspado y comprobar con microscopio o endoscopio la ausencia de restos que podrán formar un mucocele. Se debe extirpar la cortical interna de las paredes sinusales con fresa para aumentar la vascularización de los injertos.
2. Oclusión del conducto nasofrontal: evita la rehabitación por la mucosa y diseminación bacteriana. Se raspa la mucosa del conducto y se desplazan los bordes hacia la fosa nasal. Los rebordes óseos son reavivados y el conducto se ocluye con una esquirla ósea habiendo colocado previamente un injerto de aponeurosis o fascia lata.
3. Relleno de la cavidad: con tejido graso (abdominal, ilíaca, muslo) que llena toda la cavidad y estimula la proliferación de tejido fibroso. Debe ser completo. Hay algunos autores (Rohrich) no rellena la cavidad.
Su dificultad consiste en la delimitación del área de los senos frontales y la ventana ósea, secuelas estéticas, osteomielitis, lesiones durales y mucocele por persistencia de la mucosa.
Otro procedimiento descripto es la Cranealización que consiste en la ablación completa de la mucosa y pared ósea posterior de los senos frontales y en la obliteración del conducto nasofrontal. Su principal indicación son las destrucciones infecciosas o traumáticas de la pared posterior del seno frontal. En este caso, los lóbulos frontales pueden invadir el espacio libre y alcanzar la pared anterior del seno.
TRATAMIENTO ENDOSCOPICO
Históricamente, la enfermedad del seno frontal ha sido tratada de manera quirúrgica por métodos externo y de obliteración. Posteriormente se creyó que la conservación del seno frontal y permeabilidad del receso frontal y orificio frontal interno daban mejor resultado general. Esto dio lugar a la creación de procedimientos externos diseñados para eliminar el padecimiento del complejo frontoetmoidal para reestablecer drenaje.
La primera obliteración completa hecha por Riedel-Schenke en 1896, propuso la extirpación por completo de la tabla anterior y piso del seno frontal y denudar completamente la mucosa con resultados estéticos desagradables. En 1903 Killian intentó minimizar la secuela estética conservando una barra de 1 mm del borde supraorbitario. Otras modificaciones incluyeron etmoidectomía, rotación de colgajos sobre la mucosa del receso frontal y ferulización, pero fue abandonado por el elevado índice de reestenosis, necrosis del borde supraorbitario, meningitis postoperatoria y muerte.
En 1914 Lothrop, proponía una etmoidectomía intranasal y método de frontoetmoidectomía externa tipo Lynch para crear comunicación frontonasal común mediante la extirpación del piso del seno frontal, tabique intersinusal y parte superior del tabique nasal. La desventaja la falta de visualización intranasal y el colapso medial de los tejidos blandos periorbitarios con la consecuente estenosis de la comunicación nasofrontal.
Lynch realizaba una incisión periorbitaria medial, eliminación de la apófisis frontal del maxilar y lámina papirácea para eliminar el piso del seno frontal y raspar su mucosa y dejaba un tubo de drenaje nasofrontal por 10 días, a pesar de ello se producía un porcentaje importante de reestenosis y colapso de tejido orbitario
Sewall, Borden y McNaught, modificaron la técnica de Lynch-Howarth utilizando un colgajo de mucoperiostio para reepitelizar el trayecto nasofrontal y colocaban un tubo de silastic por cuatro semanas. Además conservaban la mucosa con una tasa de éxito del 97% a los seis años. Neel y Lake conservan la apófisis frontal del maxilar y el piso lateral del seno frontal para impedir el colapso orbitario. A pesar de todas las modificaciones realizadas, la tasa de fracaso a largo plazo para restablecer el drenaje del seno fue mayor al 30% lo que indicaba la necesidad de un método diferente.
Con el advenimiento de microscopios y endoscopios nasales, hubo un resurgimiento para establecer la comunicación nasofrontal por vía endonasal.
En 1991, Draf informó una experiencia de 12 años con tasas de éxito mayor de 90% en sus tres disecciones frontoetmoidales con microscopio quirúrgico. Las tres disecciones buscan restablecer la vía nasofrontal, que este limitada en todos sus lados por hueso y cubierta por mucosa para impedir el colapso medial de los tejidos blandos orbitarios, retracción cicatrizal y reestenosis.
El Draf tipo I: etmoidectomía anterior completa para que el orifico del seno frontal drene en un punto más inferior.
Draf tipo II: resección unilateral del piso del seno frontal del borde orbitario lateral a la parte anterior del tabique nasal.
Draf tipo III: extirpa la porción superior del tabique nasal, el piso del seno frontal hasta la órbita y porción inferior del tabique interfrontal. Se reserva en casos de mucoceles en situación medial o de complicaciones intracraneales en ausencia de destrucción ósea.
Las indicaciones de sinusotomía frontal endoscópica derivan de los antecedentes del enfermo, la endoscopia diagnóstica y la tomografía cortes coronales, axiales y con reconstrucción sagital helicoidal que muestra con claridad la anatomía preoperatoria. Puede usarse con éxito en la mayor parte de casos primarios de sinusitis frontal crónica, en cirugía de revisión, mucoceles y papiloma invertido del receso frontal.
La sinusitis micótica, mucoceles con erosión de la tabla posterior o pared orbitaria superior, papiloma invertido del seno frontal y senos frontales muy neumatizados son contraindicación de obliteración. Estos trastornos se atienden mejor con FESS o con una combinación de colgajo osteoplástico sin obliteración y un método intranasal donde el receso frontal y el orificio frontal interno puedan vigilarse ambulatoriamente en consultorio.
La sinusotomía frontal endoscópica esta contraindicada en.
1) tumores del seno frontal
2) papiloma invertido del seno frontal
3) osteomas de base amplia
4) estenosis del receso frontal
5) mucocele de base lateral
Todos los pacientes con sinusitis crónica deben recibir un tratamiento médico sustancial durante tres a seis semanas: antibióticos, esteroides o mucolíticos. El fracaso de un tratamiento médico extenso e individualizado para aliviar los síntomas en seis a ocho semanas justifica la tomografía para valorar la obstrucción y si se confirma se explica a los pacientes los riesgos y beneficios de FESS.
Técnica:
Debe comenzar con la revisión de las celdillas que pueden obstruir el receso frontal: Agger Nasi, etmoidales supraorbitarias y frontales que son celdillas etmoidales anteriores que neumatizan el receso frontal y lo obstruyen. Otras son las de la bulla frontal, las suprabullares y las del tabique interfrontal.
Es indispensable conservar la mucosa periférica del receso frontal y del orificio interno para evitar la reestenosis.
Primero se extirpa la apófisis unciforme y se identifica el orificio natural del seno maxilar que se deja indemne. Eliminación de la bulla etmoidal y celdillas etmoidales posteriores si hay compromiso de esa región. Si se realizara la etmoidectomía completa, debe avanzarse hasta el pico del esfenoides y base de cráneo extirpando todas las láminas. Primero deben limpiarse las celdillas etmoidales posteriores y anteriores. Al llegar al área de la arteria etmoidal, la base de cráneo se inclina hacia arriba: entrada posterior del receso frontal, aquí los movimientos se hacen de atrás hacia delante para proteger a la órbita y fosa craneal anterior. Evitar lesionar a los lados de la lámina papirácea y en sentido medial a la lámina lateral de la lámina cribosa del etmoides que es el punto más débil de toda la base de cráneo anterior lugar donde penetra la arteria etmoidal anterior. Luego con raspas anguladas se deslizan hasta la pared posterior del receso y se localiza el orificio frontal interno para eliminar la obstrucción.
En casos de revisión, retracción cicatrizal del receso o cuando hay celdillas del Agger Nasi que impidan el acceso del endoscopio, se puede realizar una trepanación externa: incisión en la porción medial de la ceja hasta llegar al periostio y con una fresa cortante de 4 mm se trepana la tabla anterior y se introduce una óptica de 30 o 70° para obtener cultivo e introducción de curetas para desobstruirlo.
La atención postoperatoria comienza en el quirófano y requiere disponer del instrumental adecuado. Hay que prevenir la retracción cicatrizal y el colapso del meato medio: en consultorio se debe debridar los coágulos de fibrina preservando la mucosa sin producir hemorragia que iniciaría de nuevo el proceso de granulación.
Para evitar el colapso del meato medio se coloca un espaciador en el meato medio durante la cirugía y se retira al cuarto día postoperatorio. Se indica al paciente lavados con solución salina para evitar la formación de costras.
Procedimiento de Lothrop modificado: es un método completamente intranasal, siendo una alternativa al tratamiento de la enfermedad crónica del seno frontal. Inicialmente se realiza una etmoidectomía para explorar el receso frontal. Tal identificación es útil pero no indispensable, ya que recordemos que puede verse dificultada por la estenosis, celdillas etmoidales supraorbitarias que se localizan en el receso frontal posterolateral donde se localiza la abertura del seno frontal en posición anteromedial. En estos casos se realiza una minitrepanación sobre el seno frontal para introducir un trócar de irrigación a través de la tabla anterior. Se administra solución salina por el trócar y bajo control endoscópico se constata la salida por el orificio natural de drenaje. Una vez localizada la región del receso frontal, se extirpa la porción superior del tabique nasal para acceder al receso frontal opuesto. También se debe resecar la porción anterior del cornete medio. Luego se extirpa la mucosa del receso frontal y el tejido blando entre los dos orificios del seno frontal y se inicia la extirpación de la separación ósea. La extirpación de hueso se dirige anterior y lateralmente para proteger el contenido intracraneal y arteria etmoidal anterior. Se elimina la separación ósea y queda una delgada cubierta de hueso en sentido anterior y lateral con amplia exposición del seno frontal. A continuación se extirpa el tabique intersinusal creándose una cavidad común: seno frontal, receso frontal y cavidad nasal conservando la mayor cantidad de mucosa sana.
Su objetivo es restaurar el aclaramiento y drenaje mucociliar al crear una gran comunicación frontonasal. Esta indicado en pacientes con sinusitis frontal crónica en los que han fracasado los procedimientos endoscópicos usuales, los que son resistentes al tratamiento médico, papiloma invertido, osteomas y mucocele del seno frontal.
No es apto cuando se presenta estrechez de la sutura nasofrontal, una profundidad estrecha anteroposterior del seno frontal o un nasión profundo que impidan el acceso intranasal y también cuando la patología crónica produce reacción con engrosamiento óseo y osteítico.
Complicaciones:
1) Fuga de LCR y meningitis: por la estrecha relación con la lámina cribosa y fosa craneal anterior.
2) Lesión del nervio óptico y tejido intraorbitario: hemorragia y pérdida de visión.
3) Lesión del saco lagrimal y epífora.
4) Hemorragia: lesión de la arteria etmoidal anterior.
Las complicaciones postoperatorias incluyen: reestenosis de la comunicación nasofrontal y recurrencia de la enfermedad. A veces se observa la estrechez circunferencial que fácilmente se extrae de forma ambulatoria en consultorio.
Marsupialización Endoscópica: tradicionalmente en EE.UU. se destaca la necesidad de la eliminación completa de la cubierta del mucocele para lograr la curación. Los que asientan en el seno frontal difieren de lesiones similares en otras cavidades porque con frecuencia el trayecto de salida del seno frontal es más estrecho y menos accesible para la cirugía intranasal. Los cirujanos han preferido los procedimientos con colgajo osteoplástico con obliteración cuya desventaja es su mayor morbilidad y dificultad en el control radiológico postoperatorio. Si el mucocele presenta expansión intracraneal el procedimiento se torna más difícil porque la cubierta esta firmemente adherida a la duramadre con el consiguiente riesgo de salida de LCR. Lo mismo ocurre si se expande al tejido blando de la frente.
La escuela europea ha realizado con buenos resultados el drenaje simple y marsupialización del mucocele con tasas de recidiva cercanas al 0%.
Los mucoceles frontales se clasifican en:
Tipo I: limitado al seno frontal (con o sin extensión orbitaria)
Tipo II: mucocele frontoetmoidal (c/s extensión orbitaria)
Tipo III: erosión de la pared posterior:
A) mínima extensión intracraneal o ninguna
B) extensión intracraneal mayor
Tipo IV: erosión de la pared anterior
Tipo V: erosión de la pared posterior y anterior:
A) mínima invasión intracraneal o sin ella
B) extensión intracraneal mayor
Técnica: como siempre es de gran importancia el estudio por imágenes y la endoscopia preoperatorio. Es preferible operar un complejo sinusal no infectado, en caso de infección se debe realizar un tratamiento antibiótico enérgico.
Se procede a la inspección del meato medio, si se extiende a ambos lados de la línea media, el cirujano decide en base de la anatomía recordando que se prefiere abrir el mucocele a través de la vía de drenaje frontal más amplia y que requiera menor trabajo óseo para ser ampliada. Se extirpa toda lesión que obstruya el receso frontal, se extirpa el Agger Nasi y si el receso drena en el infundíbulo se realiza la etmoidectomía anterior. Se localiza la arteria etmoidal anterior que se ve como una barra transversal en la base de cráneo, la abertura frontal se sitúa 2 a 4 mm. por delante de la arteria, esta distancia puede estar ocupado por celdillas etmoidales que deben extraerse. La parte posterior del receso frontal se relaciona con la fosa craneal anterior, cualquier trabajo óseo debe efectuarse en sentido anterior y en sentido posteroanterior. La sinusotomía frontal se completa al ampliar el drenaje del seno frontal en sentido anteromedial y se extirpa la escotadura nasal que forma parte del piso del seno. Se identifica el mucocele, se abre y se envía el material a cultivo, se aspira suavemente sobre todo si se expande a nivel intracraneal. Se inspecciona la cavidad del mucocele con el endoscopio pero no se raspa ni se extirpa su recubrimiento. En mucoceles con gran componente etmoidal y base amplia en al cavidad nasal, basta la marsupialización amplia sin férula. De otra manera se colocan endoprótesis en la cavidad del mucocele y su vía de salida que se fija al tabique. Se retira 6 a 12 semanas luego de la cirugía. Se indican antibióticos por 10 días y lavados con solución salina a través de la sonda, en caso de invasión intracraneal los lavados los realiza el cirujano.
Inherente al concepto de marsupialización está la capacidad del epitelio para volver a ser normal. Estudios realizados por Luna demuestran que la cubierta del mucocele no pierde características histológicas de mucosa respiratoria, incluso estudios demuestran que seis meses después de la marsupialización hay áreas de epitelio ciliado normal. Con estos datos, los autores apoyan el concepto de que no hay necesidad de eliminar por completo la cubierta del mucocele y que la FESS es el método de elección para el tratamiento de los mucoceles frontales.
A manera de resumen la Marsupialización Transetmoidal (9) es el método de elección. En los mucoceles con participación etmoidal difusa se realiza etmoidectomía amplia por vía combinada. Limitado al etmoides anterior: etmoidectomía externa o marsupialización directa a la fosa nasal.
MUCOCELE MAXILAR
Representa menos del 10% de los mucoceles, es más prevalente en Japón donde en el 50% de los casos hay antecedentes de Caldwell- Luc realizada previamente. La endoscopia permite el diagnóstico temprano del mismo mediante la siguiente tríada: abombamiento de la pared medial del seno maxilar, prolapso mucoso del meato medio con la consiguiente obstrucción del mismo y la presencia de secreción purulenta a través del meato medio. Esto se confirma con la tomografía que demuestra un seno expandido y totalmente opacificado con remodelación ósea y/o erosión. (Fig.12)
Figura 12. Mucocele maxilar con expansión del seno, remodelado y erosión ósea.
Históricamente el tratamiento de elección es la cirugía de Caldwell- Luc, remoción de la cubierta del mucocele y la confección de la contraabertura meatal inferior creando una extensa comunicación entre la lesión y la cavidad nasal.
Busaba y Salman realizan la evacuación del contenido del mucocele por vía endoscópica realizando una meatotomía media amplia que favorezca el drenaje del mismo siempre y cuando no este extendido a los tejidos blandos. No encontraron recurrencias en el lapso de 10 a 66 meses y el control de las mismas, es relativamente sencillo. Si el mucocele erosiona la tabla anterior del seno, se extiende a la fosa pterigomaxilar o si la evacuación endoscópica no fue satisfactoria, se debe recurrir a la cirugía de Caldwell-Luc. Lo mismo ocurre si su origen es traumático o consecuente a cirugías previas en el seno que hacen más dificultoso el tratamiento endoscópico.
CONCLUSIÓN
El mucocele de los senos paranasales, son formaciones pseudotumorales revestidas de epitelio pseudoestratificado o columnar bajo que se expanden lentamente y que requieren frecuentemente de años para ser sintomáticos.
Con el aumento de tamaño se extienden fuera de los límites del seno comprometido, ejerciendo efecto compresivo sobre las estructuras vecinas desplazando las partes blandas y generando reacciones de erosión o de condensación ósea con gran potencial para originar complicaciones.
La Tomografía axial computada con cortes ultrafinos y la Resonancia Magnética Nuclear son los estudios de elección para confirmar el diagnóstico. Entre los diagnósticos diferenciales se incluyen: tumores derivados de las células de Schwann, fibroma osificante, ameloblastoma, pólipo nasal, meningoencefalocele, angiofibroma, sinusitis micótica, etc.
Sin lugar a dudas el tratamiento de elección es la cirugía, asegurando un amplio drenaje sinusonasal con el objeto de evitar la recidiva que es la principal complicación en el manejo de esta patología.
Aunque la cirugía endoscópica, ha revolucionado el tratamiento de la rinosinusitis crónica, en el tratamiento del mucocele, la escuela tradicional de Estados Unidos ha enfatizado la necesidad de la remoción completa de la lesión quística incluyendo su cápsula para lograr la curación. En el caso del seno frontal la obliteración de la cavidad ha sido evocada. Sin embargo los procedimientos obliterativos presentan mayor morbilidad quirúrgica y dificultades con las imágenes radiográficas postoperatorias del seno. El drenaje simple y la marsupialización ha sido realizada por rinólogos en Europa sin ningún efecto adverso a largo plazo. El abordaje endoscópico parece ser idealmente un método radical, igualmente efectivo que los abordajes externos, pero con menor morbilidad y alteraciones anatómicas.
Aún así el tratamiento sigue siendo controversial con distintas posturas sobre si resecar o no completamente la cápsula, además requiere que el cirujano esté bien familiarizado con la técnica y conozca minuciosamente la anatomía de la región.
BIBLIOGRAFÍA ABSCESO EPIDURA INTRACRANEAL
Danner RL, Hartman BJ. Update on spinal epidural abscess: 35 cases and review of the literature. Rev Infect Dis 1987; 9:265.
Nussbaum ES, Rigamonti D, Standiford H, et al. Spinal epidural abscess: a report of 40 cases and review. Surg Neurol 1992; 38:225.
Gellin BG, Weingarten K, Gamache FW Jr, et al. Epidural Abscess. In: Infections of the Central Nervous System, 2nd Ed, Scheld WM, Whitley RJ, Durack DT (Eds), Lippincott-Raven Publishers, Philadelphia 1997. p.507.
Pradilla G, Ardila GP, Hsu W, Rigamonti D. Epidural abscesses of the CNS. Lancet Neurol 2009; 8:292.
Darouiche RO, Hamill RJ, Greenberg SB, et al. Bacterial spinal epidural abscess. Review of 43 cases and literature survey. Medicine (Baltimore) 1992; 71:369.
Akalan N, Ozgen T. Infection as a cause of spinal cord compression: a review of 36 spinal epidural abscess cases. Acta Neurochir (Wien) 2000; 142:17.
Kapeller P, Fazekas F, Krametter D, et al. Pyogenic infectious spondylitis: clinical, laboratory and MRI features. Eur Neurol 1997; 38:94.
Torda AJ, Gottlieb T, Bradbury R. Pyogenic vertebral osteomyelitis: analysis of 20 cases and review. Clin Infect Dis 1995; 20:320.
Ju KL, Kim SD, Melikian R, et al. Predicting patients with concurrent noncontiguous spinal epidural abscess lesions. Spine J 2015; 15:95.
Sørensen P. Spinal epidural abscesses: conservative treatment for selected subgroups of patients. Br J Neurosurg 2003; 17:513.
Ptaszynski AE, Hooten WM, Huntoon MA. The incidence of spontaneous epidural abscess in Olmsted County from 1990 through 2000: a rare cause of spinal pain. Pain Med 2007; 8:338.
Park KH, Cho OH, Jung M, et al. Clinical characteristics and outcomes of hematogenous vertebral osteomyelitis caused by gram-negative bacteria. J Infect 2014; 69:42.
Cook TM, Counsell D, Wildsmith JA, Royal College of Anaesthetists Third National Audit Project. Major complications of central neuraxial block: report on the Third National Audit Project of the Royal College of Anaesthetists. Br J Anaesth 2009; 102:179.
Sethna NF, Clendenin D, Athiraman U, et al. Incidence of epidural catheter-associated infections after continuous epidural analgesia in children. Anesthesiology 2010; 113:224.
Pöpping DM, Zahn PK, Van Aken HK, et al. Effectiveness and safety of postoperative pain management: a survey of 18 925 consecutive patients between 1998 and 2006 (2nd revision): a database analysis of prospectively raised data. Br J Anaesth 2008; 101:832.
Reynolds F. Neurological infections after neuraxial anesthesia. Anesthesiol Clin 2008; 26:23.
Gosavi C, Bland D, Poddar R, Horst C. Epidural abscess complicating insertion of epidural catheters. Br J Anaesth 2004; 92:294; author reply 294.
Scott DB, Hibbard BM. Serious non-fatal complications associated with extradural block in obstetric practice. Br J Anaesth 1990; 64:537.
Gaul C, Neundörfer B, Winterholler M. Iatrogenic (para-) spinal abscesses and meningitis following injection therapy for low back pain. Pain 2005; 116:407.
Darouiche RO. Spinal epidural abscess. N Engl J Med 2006; 355:2012.
Sendi P, Bregenzer T, Zimmerli W. Spinal epidural abscess in clinical practice. QJM 2008; 101:1.
Rigamonti D, Liem L, Wolf AL, et al. Epidural abscess in the cervical spine. Mt Sinai J Med 1994; 61:357.
Griffiths DL. Tuberculosis of the spine: a review. Adv Tuberc Res 1980; 20:92.
Chen WC, Wang JL, Wang JT, et al. Spinal epidural abscess due to Staphylococcus aureus: clinical manifestations and outcomes. J Microbiol Immunol Infect 2008; 41:215.
Holt HM, Andersen SS, Andersen O, et al. Infections following epidural catheterization. J Hosp Infect 1995; 30:253.
Phillips JM, Stedeford JC, Hartsilver E, Roberts C. Epidural abscess complicating insertion of epidural catheters. Br J Anaesth 2002; 89:778.
Centers for Disease Control and Prevention. Injection Safety. http://www.cdc.gov/injectionsafety/ (Accessed on June 24, 2014).
Davis DP, Wold RM, Patel RJ, et al. The clinical presentation and impact of diagnostic delays on emergency department patients with spinal epidural abscess. J Emerg Med 2004; 26:285.
Mooney RP, Hockberger RS. Spinal epidural abscess: a rapidly progressive disease. Ann Emerg Med 1987; 16:1168.
Liem LK, Rigamonti D, Wolf AL, et al. Thoracic epidural abscess. J Spinal Disord 1994; 7:449.
Curry WT Jr, Hoh BL, Amin-Hanjani S, Eskandar EN. Spinal epidural abscess: clinical presentation, management, and outcome. Surg Neurol 2005; 63:364.
Davis DP, Salazar A, Chan TC, Vilke GM. Prospective evaluation of a clinical decision guideline to diagnose spinal epidural abscess in patients who present to the emergency department with spine pain. J Neurosurg Spine 2011; 14:765.
Reihsaus E, Waldbaur H, Seeling W. Spinal epidural abscess: a meta-analysis of 915 patients. Neurosurg Rev 2000; 23:175.
Wong D, Raymond NJ. Spinal epidural abscess. N Z Med J 1998; 111:345.
Euba G, Narváez JA, Nolla JM, et al. Long-term clinical and radiological magnetic resonance imaging outcome of abscess-associated spontaneous pyogenic vertebral osteomyelitis under conservative management. Semin Arthritis Rheum 2008; 38:28.
Moseley IF, Kendall BE. Radiology of intracranial empyemas, with special reference to computed tomography. Neuroradiology 1984; 26:333.
Wheeler D, Keiser P, Rigamonti D, Keay S. Medical management of spinal epidural abscesses: case report and review. Clin Infect Dis 1992; 15:22.
Siddiq F, Chowfin A, Tight R, et al. Medical vs surgical management of spinal epidural abscess. Arch Intern Med 2004; 164:2409.
Tunkel AR, Hartman BJ, Kaplan SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis 2004; 39:1267.
Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis 2011; 52:e18.
Pfausler B, Spiss H, Beer R, et al. Treatment of staphylococcal ventriculitis associated with external cerebrospinal fluid drains: a prospective randomized trial of intravenous compared with intraventricular vancomycin therapy. J Neurosurg 2003; 98:1040.
Jorgenson L, Reiter PD, Freeman JE, et al. Vancomycin disposition and penetration into ventricular fluid of the central nervous system following intravenous therapy in patients with cerebrospinal devices. Pediatr Neurosurg 2007; 43:449.
Wang Q, Shi Z, Wang J, et al. Postoperatively administered vancomycin reaches therapeutic concentration in the cerebral spinal fluid of neurosurgical patients. Surg Neurol 2008; 69:126.
Nau R, Prange HW, Menck S, et al. Penetration of rifampicin into the cerebrospinal fluid of adults with uninflamed meninges. J Antimicrob Chemother 1992; 29:719.
Perlroth J, Kuo M, Tan J, et al. Adjunctive use of rifampin for the treatment of Staphylococcus aureus infections: a systematic review of the literature. Arch Intern Med 2008; 168:805.
von Specht M, Gardella N, Tagliaferri P, et al. Methicillin-resistant Staphylococcus aureus in community-acquired meningitis. Eur J Clin Microbiol Infect Dis 2006; 25:267.
Pintado V, Meseguer MA, Fortún J, et al. Clinical study of 44 cases of Staphylococcus aureus meningitis. Eur J Clin Microbiol Infect Dis 2002; 21:864.
Gallagher RM, Pizer B, Ellison JA, Riordan FA. Glycopeptide insensitive Staphylococcus aureus subdural empyema treated with linezolid and rifampicin. J Infect 2008; 57:410.
Kessler AT, Kourtis AP. Treatment of meningitis caused by methicillin-resistant Staphylococcus aureus with linezolid. Infection 2007; 35:271.
Naesens R, Ronsyn M, Druwé P, et al. Central nervous system invasion by community-acquired meticillin-resistant Staphylococcus aureus. J Med Microbiol 2009; 58:1247.
Ntziora F, Falagas ME. Linezolid for the treatment of patients with central nervous system infection. Ann Pharmacother 2007; 41:296.
Levitz RE, Quintiliani R. Trimethoprim-sulfamethoxazole for bacterial meningitis. Ann Intern Med 1984; 100:881.
Vartzelis G, Theodoridou M, Daikos GL, et al. Brain abscesses complicating Staphylococcus aureus sepsis in a premature infant. Infection 2005; 33:36.
Lee DH, Palermo B, Chowdhury M. Successful treatment of methicillin-resistant staphylococcus aureus meningitis with daptomycin. Clin Infect Dis 2008; 47:588.
Wallace MR, Sander AW, Licitra C, et al. Methicillin-resistant Staphylococcus aureus meningitis successfully treated with daptomycin. Infect Dis Clin Pract 2009; 17:69.
Khanna RK, Malik GM, Rock JP, Rosenblum ML. Spinal epidural abscess: evaluation of factors influencing outcome. Neurosurgery 1996; 39:958.
Baker AS, Ojemann RG, Swartz MN, Richardson EP Jr. Spinal epidural abscess. N Engl J Med 1975; 293:463.
Koo DW, Townson AF, Dvorak MF, Fisher CG. Spinal epidural abscess: a 5-year case-controlled review of neurologic outcomes after rehabilitation. Arch Phys Med Rehabil 2009; 90:512.
Heran NS, Steinbok P, Cochrane DD. Conservative neurosurgical management of intracranial epidural abscesses in children. Neurosurgery 2003; 53:893.
BIBLIOGRAFÍA SOBRE MUCOCELE DE SENOS PARANASALES:FUNCACIÓN ARAUZ OTORRINOLARINGOLOGÍA ARGENTINA
http://www.farauzorl.org.ar/mucocele-de-los-senos-paranasales