Una paciente de sexo femenino de 40 años fue internada por disminución de la agudeza visual en OI y visión doble.
La paciente había estado bien hasta 7 semanas antes del ingreso, cuando, a finales de verano, un dolor de cabeza frontal moderada desarrolló en el lado izquierdo que se resolvió completamente después de varias horas. Al despertar a la mañana siguiente, el lado izquierdo de su cara estaba adormecida desde el párpado inferior hasta la boca y de la nariz a la oreja. Ella fue evaluada en el servicio de urgencias. La tomografía computarizada (TC) de cráneo sin contraste, no reveló evidencias de hemorragia, masa, efecto de masa, u otras anomalías intracraneales. La paciente fue dada de alta. Durante los siguientes 2 semanas, la hipoestesia persistió en la mitad de la cara; fue vista por su clínico de atención primaria, un neurólogo y un dentista los cuales no revelaron ninguna anormalidad.
Dos semanas después de la aparición de los síntomas, el entumecimiento de la hemicara se resolvió pero desarrolló ptosis del párpado superior izquierdo junto con la visión doble caracterizada por una imagen que aparecía en diagonal sobre otra. La paciente consultó a otro hospital, donde se hizo una TC y una angio-TC de cabeza y cuello que reveló engrosamiento de la mucosa de los senos etmoidales y seno esfenoidal izquierdo, así como los niveles hidroaéreos en los senos maxilares bilateralmente. Un recuento sanguíneo completo, recuento diferencial de glóbulos blancos, y panel metabólico fueron normales, y una prueba de embarazo fue negativa. Se administró un curso de 2 semanas de un agente antibiótico no especificado. Varios días después, una resonancia magnética (RMN) de cráneo, después de la administración de gadolinio, al parecer reveló cambios consistentes con sinusitis, pero lo demás era normal. La agudeza visual era de 20/20 en ambos ojos, y un oftalmólogo le recetó un parche en el ojo para tratar la diplopía. Análisis de líquido cefalorraquídeo que se obtuvo por punción lumbar (incluyendo las pruebas de ácido nucleico del virus del herpes simple) y hemocultivos fueron negativos según los informes.
Dos semanas antes de la internación, comenzó con dolor periorbitario en el lado medial izquierdo, sin presión, dolor de cabeza, o visión doble. Una semana más tarde, repitió diplopia, con desviación anormal asociada del ojo izquierdo. Los resultados de estudios de potenciales evocados visuales eran no diagnósticos. En el ojo izquierdo, la agudeza visual se redujo y la discriminación para color era anormal. Tres días antes de su ingreso, la paciente tuvo náuseas y vómitos, junto con vértigo que dio lugar a caída en su cuarto de baño. La diplopía persistió, y el dolor periorbitario izquierdo era de 5 en una escala de 0 a 10 (con 10 indicando el dolor más severo). La noche antes de la internación se repitió la RMN en el segundo hospital.7
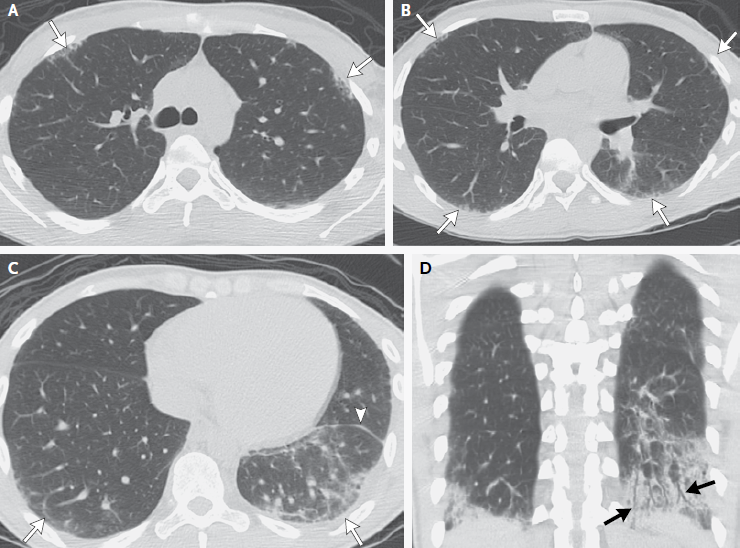
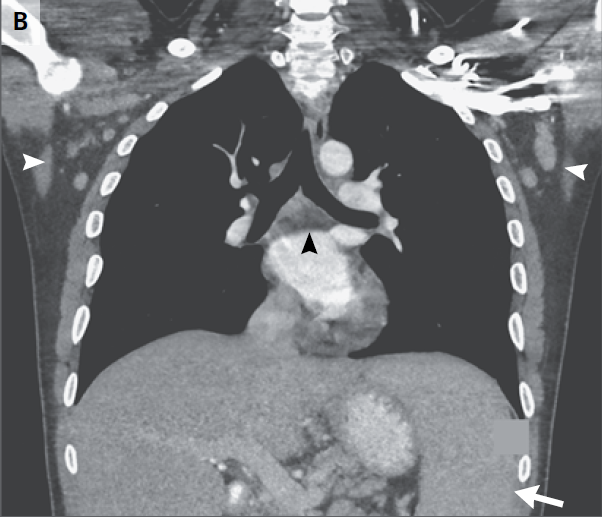
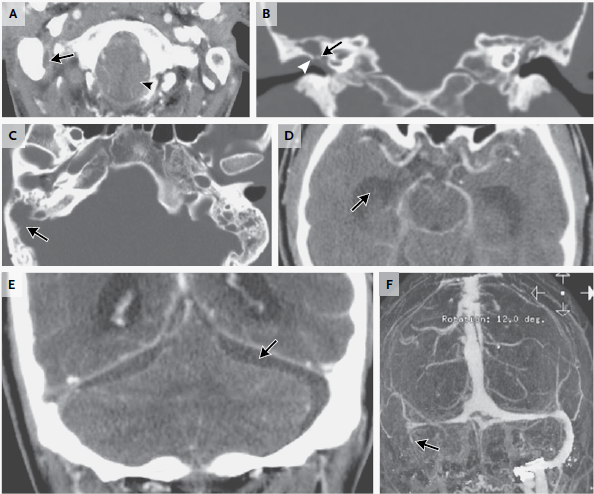
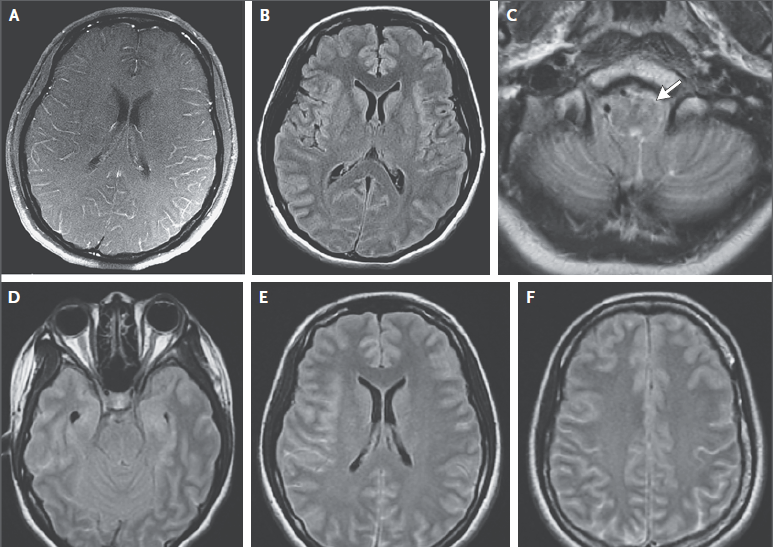
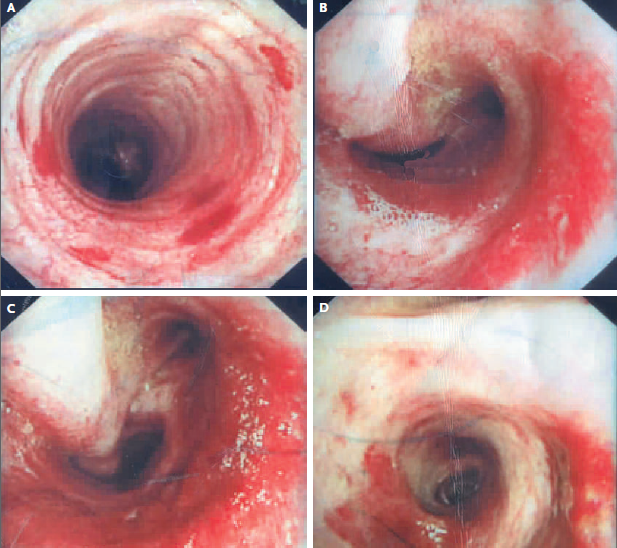
RMN de cráneo mostró tejido blando anormal a lo largo de las paredes anterior y lateral del seno esfenoidal izquierdo, en el vértice de la órbita izquierda extendiéndose posteriormente en el seno cavernoso izquierdo. Este tejido blando era de densidad intermedia tanto en una imagen potenciada en T2 (Figura A) Y una imagen potenciada en T1 con contraste (Figura 1B). Una imagen de RMN con contraste, coronal (Figura 1C) mostró aumento anormal en la unión del seno cavernoso y la cavum de Meckel en el lado izquierdo, con realce de tejido que se extendía a la parte interna del foramen oval.
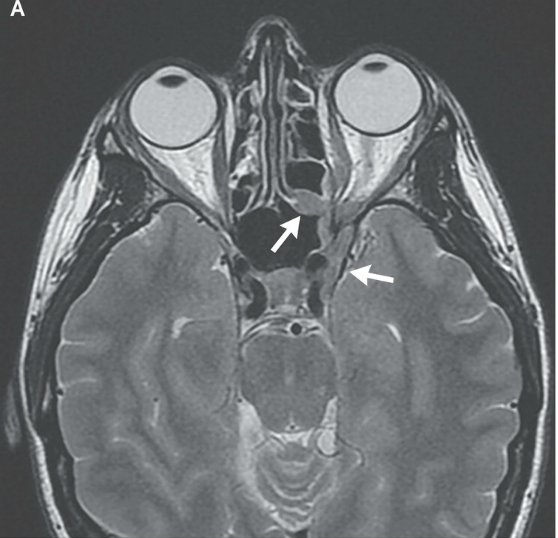
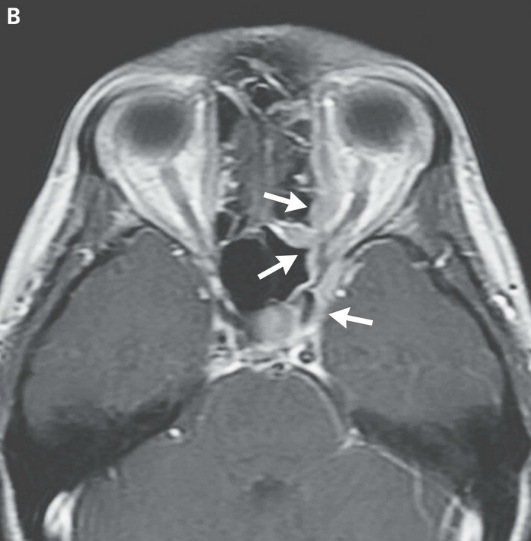


Figura 1
Estudios por imágenes de los senos paranasales y órbitas.
RMN DE ÓRBITAS REALIZADA EN OTRO HOSPITAL. Una imagen en T2 (Panel A)y una T1 con contraste (Panel B) muestra tejido de densidad intermedia tapizando el seno esfenoidal y afectando el vértice de la órbita izquierda y el seno cavernoso (flechas). Un corte coronal en T1 (Panel C) muestra realce de partes blandas extendiéndose inferiormente al foramen oval. Una TC de órbita (Panel D) muestra que el hueso entre el seno esfenoidal y el vértice de la órbita están relativamente intactos a pesar de la presencia de tejido con densidad de partes blandas anormal a ambos lados (flecha).
Sobre la base de los resultados de la resonancia magnética, se informó a la paciente a acudir de inmediato al servicio de urgencias de otro hospital. A su llegada, se informó que el ibuprofeno proporcionado generó alivio mínimo del dolor periorbitario. La paciente no tenía antecedentes de fiebre, sudores nocturnos, dolor en el pecho, disuria, diarrea, erupciones cutáneas, dolores musculares o articulares, o pérdida de peso.
Tenía antecedentes de hipotiroidismo, rinosinusitis crónica y alergias ambientales a mohos y polvo; ella había tenido herpes zoster que afectó el nervio trigémino que se había resuelto y tenía antecedente de una cirugía de un fibroadenoma de mama en el pasado. Los medicamentos que tomaba eran levotiroxina, aspirina, un suplemento de calcio con vitamina D, e ibuprofeno para el dolor. Ella no tenía alergias a los medicamentos. Vivía con su marido y sus hijos y trabajaba desde su casa. Ella bebía alcohol de vez en cuando y no fumaba ni usaba drogas ilícitas. Su abuelo materno había tenido linfoma; no se conocía otra historia familiar.
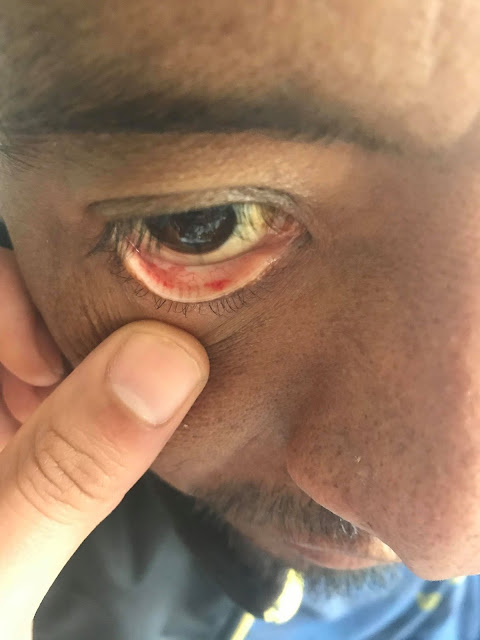
En el examen, la temperatura era de 37,4 ° C; los otros signos vitales y la saturación de oxígeno eran normales. El examen oftalmológico reveló una agudeza visual de 20/20 en el ojo derecho y 20/400 en el ojo izquierdo. Las pupilas eran iguales, redondas y reactivas, con un defecto pupilar aferente izquierdo. Resultados de las pruebas de color visión con placas de color de Ishihara fueron normales en el lado derecho (8/8 correctamente identificados) y 0/8 de la izquierda. La presión intraocular era 13 mm Hg en el lado derecho y 11 mm Hg a la izquierda. Examen con lámpara de hendidura reveló segmentos normales anteriores bilateralmente. El examen de la motilidad extraocular reveló plena movilidad en el ojo derecho; el ojo izquierdo tenía aducción limitada, supraducción y infraducción, con intorsion mínimo en intento de secuestro (Figura 2 ).
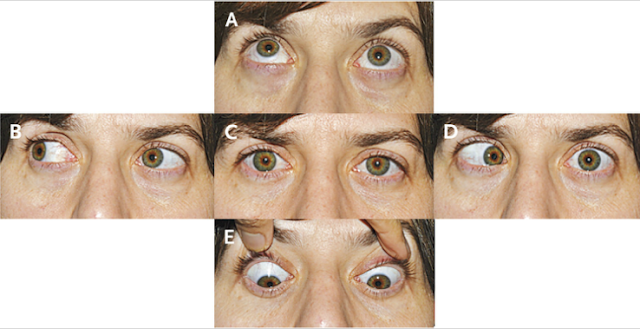
Figura 2
Fotografías de los ojos del paciente.
En la mirada primaria (Panel C) hay leve ptosis del párpado superior izquierdo.El ojo izquierdo tiene leve déficit de elevación (Panel A), aducción (Panel B), y depresión (Panel E), y no tiene abeducción (PanelD).
Había leve disminución de la sensibilidad al pinchazo en la segunda rama del trigémino y la fuerza de la hemicara era normal. Resultados del examen de fondo de ojo fueron normales bilateralmente; los nervios ópticos fueron normales, sin palidez, edema, o hemorragias. Resultados de las pruebas de campo visual, fueron normales en el lado derecho; el lado izquierdo tenía un gran escotoma central, con la preservación de la visión periférica. El examen endonasal endoscópico reveló que los cornetes fueron moderadamente hipertrofiado; un espolón óseo septal izquierdo y desviación septal leve a moderada estaban presentes. El resto de los, exámenes generales ,neurológicos y otorrinolaringológicos eran normales.
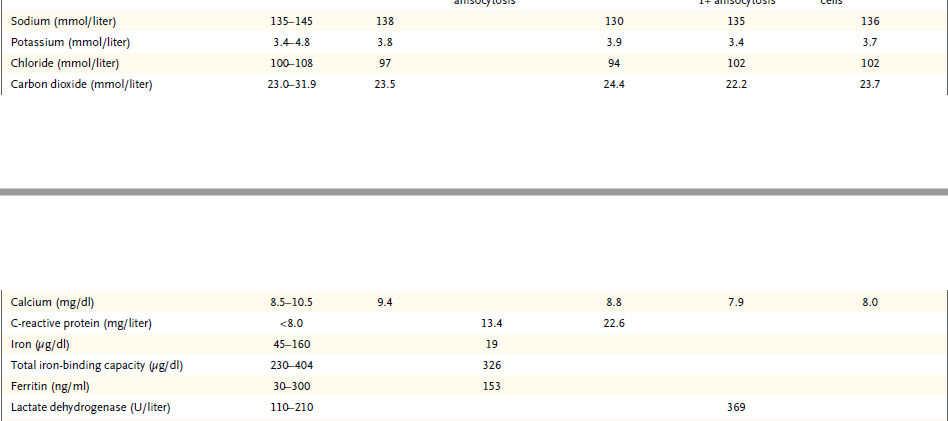
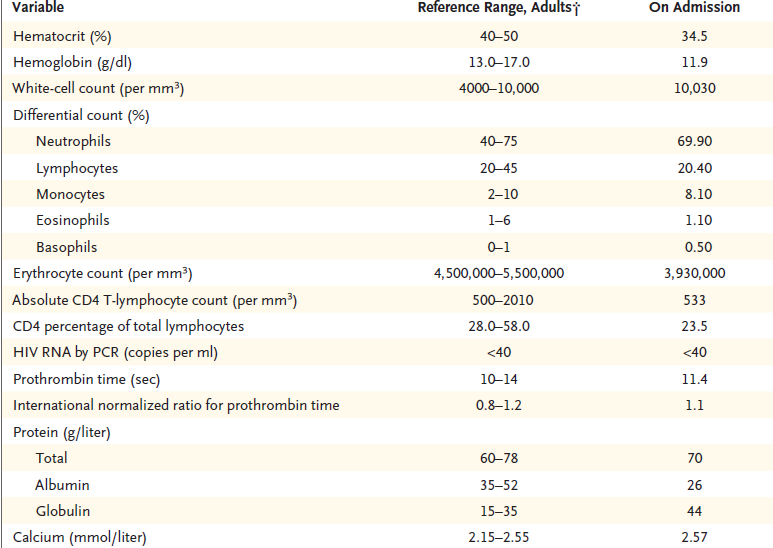
El hematocrito, hemoglobina, recuento de glóbulos blancos, recuento diferencial, recuento de plaquetas, y velocidad de sedimentación globular fue normal, al igual que los niveles sanguíneos de electrolitos, calcio, fósforo, magnesio, glucosa, proteína C reactiva, la enzima convertidora de la angiotensina, y inmunoglobulinas (IgG, IgA e IgM) y los resultados de la función renal y pruebas de coagulación. Las pruebas para la gonadotropina coriónica humana en la sangre y en la orina fue negativo. Pruebas de suero fue positivo para anticuerpos anti-Ro (unidades de densidad óptica, 63,37; intervalo de referencia, 0-19,99) y anticuerpos antinucleares (en 1:40 y 1: 160 diluciones, en un patrón homogéneo); las pruebas de anticuerpos citoplásmicos antineutrófilos (anti-PR3 y anti-MPO), anti-La y anticuerpos anti ADN de doble cadena fue negativo. El análisis de orina reveló 1+ cetonas, traza de sangre oculta y proteínas, con 0-2 células rojas, muy pocas células tubulares renales, y pocas células escamosas por campo de alto poder y mucina por campo de bajo consumo. La paciente fue ingresada en el hospital.
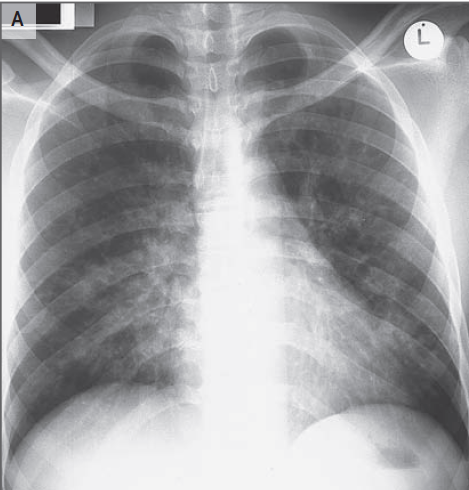
En el segundo día, una punción lumbar reveló una presión de apertura de 17,5 cm de agua. El análisis del líquido cefalorraquídeo reveló muy pocas células mononucleares y no había glóbulos blancos o rojos. Los niveles de glucosa, proteína total, y la enzima convertidora de angiotensina fueron normales; pruebas para la lipoproteína de muy baja densidad fue negativa. Tinción de Gram no reveló ningún organismo, la electroforesis no reveló bandas oligoclonales, y el examen citológico no reveló células malignas. Una radiografía de tórax mostró pulmones claros y sin evidencia de adenopatías mediastínicas. En el tercer día, se realizó TC de las órbitas.
TC de las órbitas (Figura 1D) reveló que el hueso entre el tejido blando anormal en el seno esfenoidal izquierdo y el tejido blando anormal en el vértice de la órbita izquierda estaba prácticamente intacto.
En el cuarto día, se llevó a cabo un procedimiento diagnóstico.
DIAGNÓSTICO DIFERENCIAL
Esta mujer 41 años de edad, tuvo déficits neurológicos intermitentes durante varias semanas. Los resultados reportados sugieren la participación de varios nervios craneales, incluyendo el segundo (asociado con disminución de la agudeza visual, discromatopsia, defecto pupilar aferente, y alteración del del campo visual), tercer nervio craneal ( ptosis y diplopía), la segunda rama del quinto nervio craneal (hipoestesia de la mejilla), y sexto ( déficit de abducción). Los estudios de imagen confirmaron que estos hallazgos estaban localizados en el seno cavernoso y vértice de la órbita.
La caracterización cuidadosa de los cambios radiográficos es importante en la determinación del diagnóstico en este caso. La enfermedad de los senos paranasales es mínima. Una masa en el vértice de la órbita izquierda que afecta al músculo recto interno está presente, con el hueso intacto entre esta área y una cámara de aire etmoidal opacificada adyacente. La masa infiltrativa se extiende hacia el seno cavernoso, duramadre, fosa pterigopalatina, fisura orbitaria inferior, y el canal óptico. En esta paciente, el diagnóstico diferencial de una masa en el vértice de la órbita con infiltración a través de múltiples agujeros y en las estructuras circundantes es amplio; sin embargo, el diagnóstico más crítico y que debe ser investigado con prontitud, es lamucormicosis.
MUCORMICOSIS
La mucormicosis es una infección por hongos potencialmente mortal que ocurre con mayor frecuencia en personas inmunocomprometidas. Las personas afectadas pueden presentar inflamación periorbitaria, oftalmoplejía externa, y pérdida visual grave como consecuencia de la destrucción angioinvasiva del tejido. La mucormicosis se origina en la nariz o los senos paranasales, aunque algunos pacientes pueden tener cambios mínimos en la radiografía de los senos paranasales. Todos los pacientes con esta constelación de hallazgos, incluyendo aquellos que se cree que son inmunocompetentes, deben someterse a una evaluación endoscópica inmediata de la nariz y los senos de tejido necrótico. Si es muy sospechoso el diagnóstico de mucormicosis, se debe realizar la biopsia y cultivo, y la terapia antifúngica debe iniciarse antes de los resultados de las pruebas.1
El diagnóstico de mucormicosis es poco probable en esta paciente porque era inmunocompetentes y el examen endoscópico no reveló ninguna evidencia de necrosis de los tejidos. Por lo tanto, un diagnóstico diferencial más amplio incluyendo infecciones, enfermedades inflamatorias y cáncer, deben ser considerados.
INFECCIÓN
La celulitis orbitaria comúnmente resulta de una infección bacteriana, más a menudo como una extensión del etmoides o sinusitis frontal, pero también puede resultar de un trauma cutáneo, absceso dentario, o dacriocistitis. Los organismos más comúnmente asociados con la celulitis orbitaria son estreptococos y estafilococos .2 Esta paciente tenía antecedentes de sinusitis crónica, y niveles de aire líquido estaban presentes en el seno en exploraciones recientes. La progresión de la enfermedad puede haber disminuido con el transcurso de los antibióticos orales que se administraron 5 semanas antes de la presentación, lo que sugiere una posible causa bacteriana. Sin embargo, ella no tenía signos externos de infección, tales como edema de párpados, eritema o quemosis conjuntival. Además, estaba afebril y tenía un recuento de glóbulos blancos y recuento diferencial normal y su evolución clínica fue indolente para una infección bacteriana aguda, particularmente una infección que involucra el seno cavernoso y la duramadre.
En casos raros, Mycobacterium tuberculosis u otras micobacterias atípicas pueden causar una infección orbitaria indolente. La tuberculosis es poco probable debido a que el paciente no tenía exposiciones conocidas o viajes al extranjero. Sin embargo, los pacientes con tuberculosis que involucran la órbita pueden presentarse con un granuloma en el vértice de la órbita como resultado de diseminación hematógena o extensión de la cavidad nasal o sinusal. La tuberculosis puede ocurrir en forma aislada o en asociación con enfermedad pulmonar. En este caso, no hay ningún registro de las pruebas de la tuberculosis después de haber sido realizadas en esta paciente, pero ella no mostró síntomas sistémicos que sugiriesen esta enfermedad (por ejemplo, tos, fiebre, o sudores nocturnos) y la radiografía de tórax fue normal.
TRASTORNOS INFLAMATORIOS
La enfermedad inflamatoria de la órbita es común, y sus causas incluyen inflamación orbitaria idiopática, inflamación orbitaria relacionadas IgG4, sarcoidosis, Granulomatosis de Wegener, y trastornos de proliferación de histiocitos.
La inflamación idiopática orbitaria, que es, con mucho, la más común de estas enfermedades, se conocía anteriormente como seudotumor orbitario y se refiere a la inflamación que involucra alguna estructura de la órbita. Nomenclatura descriptiva específica incluye dacrioadenitis, escleritis y miositis, aunque muchos casos implican la infiltración difusa de la grasa orbitaria. La enfermedad de esta paciente implica no sólo la grasa del vértice de la órbita, sino también el músculo recto medial. Esta condición suele ser dolorosa a menudo resulta en inflamación periorbitaria visible y en ocasiones puede extenderse a la participación de los senos paranasales o la dura; estas características se observaron en esta paciente.5 La presencia de dolor suele ser clínicamente útil para hacer el diagnóstico, pero la ausencia de dolor pueden ser engañosas. Esta paciente tuvo episodios de dolor durante el curso evolutivo de la enfermedad. Su primer síntoma fue un dolor de cabeza frontal en el lado izquierdo. Entonces, después de varias semanas de déficits neurológicos crecientes y menguantes, desarrolló dolor periorbitario medial izquierdo y persistió durante las 2 semanas antes de la admisión. Los resultados normales de análisis de líquido cefalorraquídeo, las imágenes del tórax, y los estudios serológicos también apoyarían este diagnóstico.
La enfermedad relacionada con IgG4 es un trastorno inflamatorio orbitario que es menos común que la inflamación orbitaria idiopática. Los pacientes con enfermedades relacionadas con IgG4 tienen presentaciones clínicas y radiológicas que son similares a las de los pacientes con inflamación orbitaria idiopática, pero la enfermedad relacionada con IgG4-es más probable que sea bilateral y asociada con un trastorno inflamatorio de otro órgano o sistema orgánicos.6 La sarcoidosis es una enfermedad granulomatosa que puede afectar los pulmones, el hígado, el bazo, los ojos y la órbita. La sarcoidosis orbitaria más a menudo involucra las glándulas lagrimales, pero puede involucrar otras estructuras orbitales y se extienden a través de agujeros apicales de las estructuras circundantes, un patrón que se ha visto en esta paciente.7 Sin embargo, la ausencia de participación de la glándula lagrimal, la ausencia de síntomas respiratorios, y hacer este diagnóstico improbable en este caso la radiografía de tórax normal. La granulomatosis de Wegener es una vasculitis necrotizante multisistémica que comúnmente implica los senos paranasales, vías respiratorias y los riñones. La uveítis es más común que la enfermedad orbital, que puede manifestarse por un granuloma focal o cambios orbitales difusas. Necrosis escleral y ayuda sinusitis destructiva para distinguir esta enfermedad de condiciones similares y no estuvieron presentes en esta paciente 8
CÁNCER
Una variedad de tipos de cáncer podrían causar una lesión en la órbita de esta paciente y las estructuras circundantes. Sus síntomas comenzaron con hallazgos neurológicos que eran compatibles con múltiples anomalías de los nervios craneales y localmente compromiso del seno cavernoso y el vértice de la órbita. Los estudios iniciales de neuroimágenes fueron bastante normales y no mostraron participación sinusal visto en las exploraciones posteriores, lo que sugiere que este proceso probablemente no se originó principalmente en el seno. Por lo tanto, los tumores del seno que tienen una propensión a la invasión orbital (como el carcinoma nasofaríngeo, carcinoma de células escamosas y el melanoma), son diagnósticos poco probables en esta paciente.
El espectro de posibles cánceres orbitales se puede reducir si se especifican en este caso debido a la naturaleza infiltrativa de la lesión. Los tumores linfoides son tipos comunes de cáncer infiltrante orbitarios y van desde la variedad más común, linfomas indolentes asociados a tejido de la mucosa , a las variedades más raras y agresivas. El linfoma puede involucrar cualquier estructura orbitaria (comúnmente incluyen la glándula lagrimal, los músculos extraoculares, o la grasa), y puede ser parte de un proceso sistémico. Linfomas de células B son el tipo más común de los que involucran a la órbita y tienden a ser unilaterales, sin dolor, y de lento crecimiento.9 En la radiografía, el linfoma tiene un patrón infiltrativo, con moldeado a las estructuras circundantes; estas características son similares al patrón descrito en esta paciente. Sin embargo, la progresión de la enfermedad bastante rápida, durante un período de 5 semanas, argumenta en contra de este diagnóstico.
El cáncer metastásico podría causar una lesión en el vértice de la órbita. Los cánceres metastásicos más comunes en adultos son carcinoma de la mama, próstata, pulmón, riñón, o del tracto gastrointestinal (incluyendo un tumor carcinoide) y melanoma. Aproximadamente el 20% de los pacientes con cáncer metastásico orbital tiene ningún tumor maligno primario conocido. Las lesiones metastásicas menudo crecen rápidamente, producen dolor, y están bien circunscritas en la radiografía, mientras que el cáncer de mama puede ser más difuso y crece a lo largo de los planos fasciales.10 Un cáncer de mama puede causar enoftalmos debido a la fibrosis y contracción de los tejidos.11 hallazgos radiográficos de este paciente y el curso temporal de la enfermedad podría ser consistente con cáncer de mama metastásico, aunque ella es una paciente sin antecedentes de cáncer de mama.
Los tumores de la vaina del nervio óptico pueden producir cambios radiográficos en el vértice de la órbita. Los meningiomas de la vaina del nervio óptico nacen de la capa aracnoides de las meninges y ocurren más comúnmente en mujeres de mediana edad. Sin embargo, sería de esperar una lesión de la vaina del nervio óptico que siga el curso del nervio óptico, sin la ramificación en los distintos agujeros que fue visto en este caso.
Las malformaciones vasculares de la órbita son muy comunes, y las malformaciones venolinfática pueden manifestarse en forma de lesiones apicales como irregular. Sin embargo, este tipo de malformaciones raras veces comprimen el nervio óptico o extienden en agujeros circundantes. Este tipo de lesión es poco probable que crezca en tan poco tiempo a menos que estuviera presente una trombosis, lo que podría dar lugar a dolor, proptosis, y característico conclusiones13 radiográfica
En conclusión, el diagnóstico más probable en este paciente es linfoma. El aspecto radiológico de la lesión de esta paciente es muy típico de linfoma, y tanto la ausencia de signos externos periorbitarios como la propagación de la enfermedad a través de varios agujeros son consistentes con este diagnóstico. Debido a la presencia de dolor y crecimiento bastante rápido, sospecho que el linfoma de este paciente es más agresivo que un típico linfoma del tejido linfoide asociado a la mucosa.
DIAGNOSTICO CLINICO PRESUNTIVO
LINFOMA.
DISCUSIÓN PATOLÓGICA
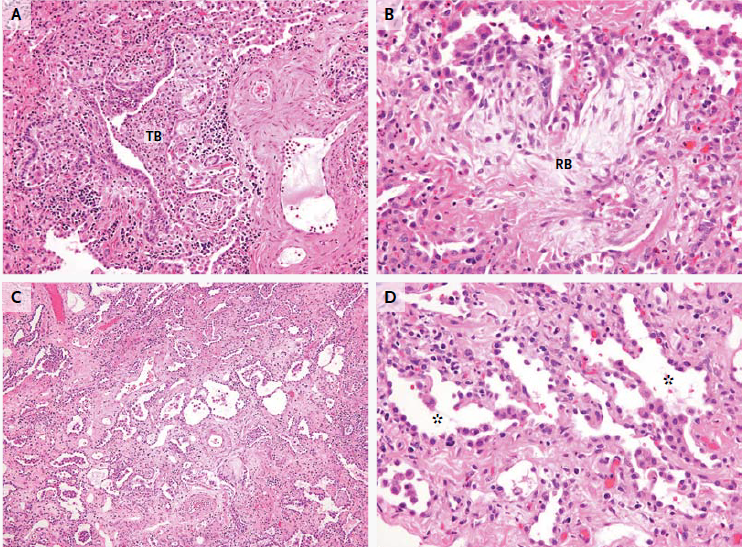
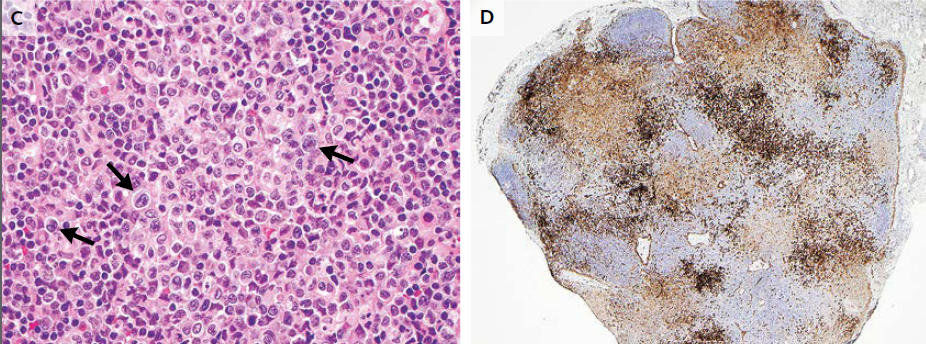
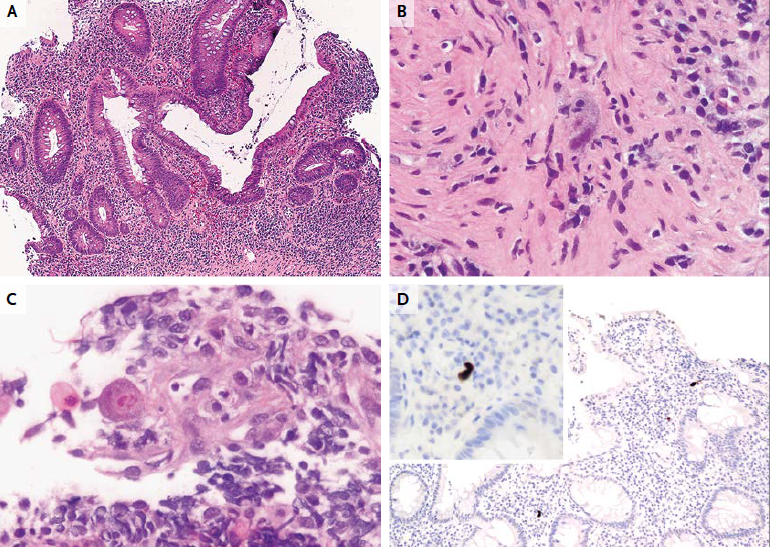
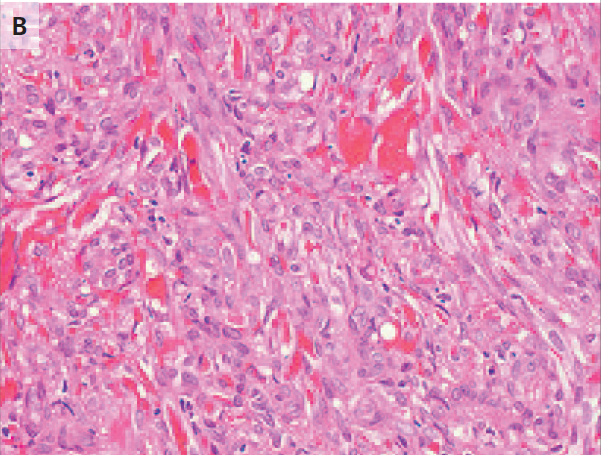
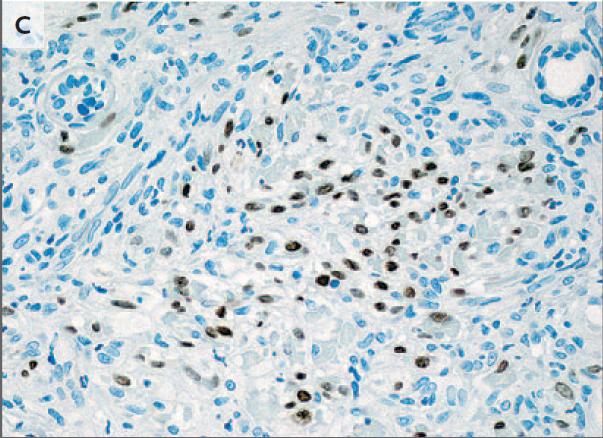
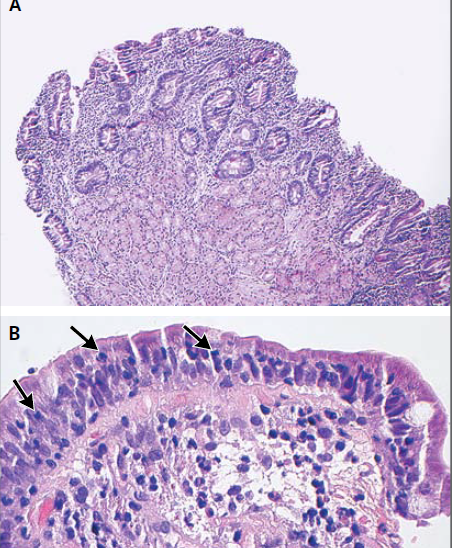
El examen de las muestras de biopsia de la etmoidal posterior izquierda y senos esfenoidales mostraron mucosa nasosinusal con un tejido linfoide denso subyacente en un fondo de la fibrosis. Las células linfoides eran grandes, y la tinción inmunohistoquímica para CD20, un antígeno de células B, fue positivo, lo que confirma el diagnóstico de linfoma difuso de células B grandes (Figura 3A, 3B y 3C).

Figura 3
Biopsia muestras de la Izquierda Posterior etmoides y esfenoides senos
En la tinción con hematoxilina y eosina, las muestras de biopsia del etmoides posterior izquierda y senos esfenoidales revelaron mucosa nasosinusal con un tejido linfoide subyacente infiltrante en un fondo de fibrosis moderada (Panel A). Las células linfoides eran grandes, con núcleos irregulares multilobulados, cromatina vesicular y nucleolos prominentes ocasionales (Grupo B). La tinción inmunohistoquímica para CD20 fue difusamente positiva (Grupo C), lo que confirma el diagnóstico de linfoma difuso de células B. En la tinción inmunohistoquímica adicional, las células tumorales fueron difusamente positiva para CD10 (Panel D) y CD30 (Panel E), y la mayoría de las células tumorales eran de forma variable positiva para el linfoma de células B 2 proteínas (BCL2) (Panel F).
El linfoma difuso de células B representa un grupo de cánceres biológicamente heterogéneos que se pueden dividir morfológica, genética e inmunofenotípicamente y que incluyen ciertas entidades específicas.14 La mayoría de los casos no cumplen los criterios diagnósticos de una de las entidades patológicas específicas y son clasificados como linfoma difuso de células B. Un examen más detallado inmunofenotípico del tumor de esta paciente reveló que las grandes células B fueron positivas para antígenos del centro germinal CD10 y linfoma de células B 6 proteínas (BCL6) y fueron negativos para el post-germinal centro mieloma múltiple marcador oncogén 1 (MUM1, también conocido como factor regulador de interferón 4), los hallazgos que indican que el linfoma es de origen germinal-centro, en lugar de origen nongerminal-centro (Figura 3D) 0,15 mayoría de las células tumorales fueron positivas para la proteína antiapoptótica marcador linfoma de células B (2 BCL2) (Figura 3F), 50% de las células tumorales fueron positivas para el antígeno de proliferación Ki-67, y 10% de las células tumorales fueron positivas para MYC. La ausencia de una alta expresión concurrente de BCL2 y MYC indica que este tumor está en un subgrupo de linfoma difuso de células B que se asocia con un mejor pronóstico, en comparación con el subgrupo de tumores que muestran alta expresión concurrente de MYC y BCL2.16 , 17 Las células tumorales fueron también fuertemente y de forma difusa positivas para CD30, un antígeno de activación (Figura 3E); un ARN en el ensayo de hibridación in situ fue negativo para el virus de Epstein-Barr.
La expresión de CD30 se ve en sólo el 14% de los casos de linfoma difuso de células B, y los casos CD30-positivas se han notificado a ser asociado con un superior global a 5 años y la supervivencia libre de progresión, en comparación con los casos-CD30 negativo, una diferencia que se mantiene en tanto germinal de centro y de centro-nongerminal subgrupos. La expresión de genes de perfiles de los estudios han demostrado un perfil distinto, lo que sugiere que los casos de CD30-positivo puede representar un subgrupo distinto de grandes linfomas difusos de células B.18
DISCUSIÓN DE MANEJO
Esta paciente tenía el estadio IA de linfoma difuso de células B que involucraba la órbita. En el tratamiento de esta enfermedad, es importante determinar si la lesión fue intradural o extradural y considerar el papel de la terapia dirigida al sistema nervioso central. En pacientes con linfoma de células B grandes sistémico difuso, la participación secundaria del sistema nervioso central se asocia con un mal pronóstico, con una supervivencia global media de 6 meses. Después de una cuidadosa consulta con el servicio de neuro-oncología y la revisión de los estudios de imagen del paciente, no se encontraron pruebas directas de la participación del sistema nervioso central. Sin embargo, estudios adicionales (incluyendo una punción lumbar para la prueba citológica, citometría de flujo, y el gen de reordenación de IgH mediante la reacción en cadena de la polimerasa )eran necesarios para descartar afectación intradural, y todas estas pruebas fueron negativas. Por lo tanto, esta paciente tenía linfoma difuso de células B en estadio IA con un alto riesgo de recaída por afectación del sistema nervioso central. La terapia estándar para esta enfermedad consiste en seis ciclos de rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona (R-CHOP). La profilaxis óptima para la afectación del sistema nervioso central recidivante en pacientes con linfoma difuso de células B aún no se ha definido. Sin embargo, ya sea metotrexato intratecal o sistémica tiene la mayor actividad contra los linfomas que implica el sistema nervioso central. El metotrexato sistémico tiene un beneficio potencial sobre metotrexato intratecal porque se asocia con una penetración más profunda en el parénquima cerebral y una cobertura más uniforme del sistema nervioso central, y se puede combinar de manera segura con R-CHOP. Debido a la ubicación del linfoma de este paciente resultó en un aumento significativo del riesgo de recaer participación del sistema nervioso central, hemos tratado este paciente con seis ciclos de R-CHOP y metotrexato sistémico fue administrada en el día 15 de cada ciclo de 21 días.
Esta paciente tuvo una respuesta completa a seis ciclos de R-CHOP. Estudios anteriores han sugerido que la administración de radioterapia de consolidación después de CHOP tiene un beneficio mínimo, pero como rituximab se ha añadido a la pauta de quimioterapia, varios estudios retrospectivos han demostrado mejores tasas de supervivencia libre de enfermedad y global entre los pacientes con células B grandes difuso linfoma que recibieron radioterapia después de R-CHOP, en comparación con aquellos que no recibieron terapia de radiación.19-22 Dado que este paciente tenía enfermedad extraganglionar localizado en la órbita, pensamos que la radioterapia adyuvante se indicó.
La terapia de protones fue seleccionado para reducir la exposición a los tejidos normales; en este paciente que había recibido altas dosis de metotrexato, fumos especialmente cuidadosos para limitar la exposición al cerebro. Una dosis de 30 Gy fue administrada. Tal tratamiento debería resultar en efectos tóxicos mínimos, como el daño a la órbita ósea, retina, nervio óptico, glándula lagrimal, y los tejidos blandos rara vez se ve con dosis de menos de 40 Gy.25 La radioterapia con dosis superiores a 12 Gy puede dar lugar a formación de cataratas 25; Por lo tanto, este paciente debe ser monitoreada para este efecto tóxico retrasado a partir de 2 a 3 años después del tratamiento.
DIAGNÓSTICO ANATÓMICO
LINFOMA DIFUSO DE CÉLULAS B GRANDES (NO ESPECIFICADO).
Traducción de:
“A 41-Year-Old Woman with Decreased Vision in the Left Eye and Diplopia”
Suzanne K. Freitag, M.D., Mary E. Cunnane, M.D., Michael K. Yoon, M.D., Jeffrey A. Barnes, M.D., Ph.D., Karen M. Winkfield, M.D., Ph.D., and Aliyah R. Sohani, M.D.
N Engl J Med 2015; 372:2337-2345June 11, 2015DOI: 10.1056/NEJMcpc141517
References
1. Warwar RE, Bullock JD. Rhino-orbital-
cerebral mucormycosis: a review. Orbit
1998; 17: 237-45.
2. Tovilla-Canales JL, Nava A, Tovilla y
Pomar JL. Orbital and periorbital infections.
Curr Opin Ophthalmol 2001; 12: 335-41.
3. Madge SN, Prabhakaran VC, Shome
D, Kim U, Honavar S, Selva D. Orbital tuberculosis:
a review of the literature. Orbit
2008; 27: 267-77.
4. Yuen SJ, Rubin PA. Idiopathic orbital
inflammation: distribution, clinical features,
and treatment outcome. Arch Ophthalmol
2003; 121: 491-9.
5. Mahr MA, Salomao DR, Garrity JA.
Inflammatory orbital pseudotumor with
extension beyond the orbit. Am J Ophthalmol
2004; 138: 396-400.
6. Ginat DT, Freitag SK, Kieff D, et al.
Radiographic patterns of orbital involvement
in IgG4-related disease. Ophthal
Plast Reconstr Surg 2013; 29: 261-6.
7. Mavrikakis I, Rootman J. Diverse clinical
presentations of orbital sarcoid. Am J
Ophthalmol 2007; 144: 769-75.
8. Muller K, Lin JH. Orbital granulomatosis
with polyangiitis (Wegener granulomatosis):
clinical and pathologic findings.
Arch Pathol Lab Med 2014; 138: 1110-4.
9. Cockerham GC, Jakobiec FA. Lymphoproliferative
disorders of the ocular adnexa.
Int Ophthalmol Clin 1997; 37: 39-59.
10. Shields JA, Shields CL, Brotman HK,
Carvalho C, Perez N, Eagle RC Jr. Cancer
metastatic to the orbit: the 2000 Robert
M. Curts Lecture. Ophthal Plast Reconstr
Surg 2001; 17: 346-54.
11. Günalp I, Gündüz K. Metastatic orbital
tumors. Jpn J Ophthalmol 1995; 39:
65-70.
12. Shapey J, Sabin HI, Danesh-Meyer HV,
Kaye AH. Diagnosis and management of
optic nerve sheath meningiomas. J Clin
Neurosci 2013; 20: 1045-56.
13. Rootman J, Heran MK, Graeb DA.
Vascular malformations of the orbit: classification
and the role of imaging in diagnosis
and treatment strategies. Ophthal
Plast Reconstr Surg 2014; 30: 91-104.
14. Stein H, Warnke RA, Chan WC. Diffuse
large B-cell lymphoma, not otherwise
specified. In: Swerdlow SH, Campo
E, Harris NL, et al., eds. WHO classification
of tumours of haematopoietic and
lymphoid tissues. Lyon, France: International
Agency for Research on Cancer Press,
2008: 233-7.
15. Hans CP, Weisenburger DD, Greiner
TC, et al. Confirmation of the molecular
classification of diffuse large B-cell lymphoma
by immunohistochemistry using
a tissue microarray. Blood 2004; 103: 275-
82.
16. Johnson NA, Slack GW, Savage KJ, et
al. Concurrent expression of MYC and
BCL2 in diffuse large B-cell lymphoma
treated with rituximab plus cyclophosphamide,
doxorubicin, vincristine, and
prednisone. J Clin Oncol 2012; 30: 3452-9.
17. Green TM, Young KH, Visco C, et al.
Immunohistochemical double-hit score is
a strong predictor of outcome in patients
with diffuse large B-cell lymphoma treated
with rituximab plus cyclophosphamide,
doxorubicin, vincristine, and prednisone.
J Clin Oncol 2012; 30: 3460-7.
18. Hu S, Xu-Monette ZY, Balasubramanyam
A, et al. CD30 expression defines a
novel subgroup of diffuse large B-cell lymphoma
with favorable prognosis and distinct
gene expression signature: a report
from the International DLBCL Rituximab-
CHOP Consortium Program Study. Blood
2013; 121: 2715-24.
19. Phan J, Mazloom A, Medeiros LJ, et al.
Benefit of consolidative radiation therapy
in patients with diffuse large B-cell lymphoma
treated with R-CHOP chemotherapy.
J Clin Oncol 2010; 28: 4170-6.
20. Dorth JA, Chino JP, Prosnitz LR, et al.
The impact of radiation therapy in patients
with diffuse large B-cell lymphoma
with positive post-chemotherapy FDG-PET
or gallium-67 scans. Ann Oncol 2011; 22:
405-10.
21. Marcheselli L, Marcheselli R, Bari A,
et al. Radiation therapy improves treatment
outcome in patients with diffuse
large B-cell lymphoma. Leuk Lymphoma
2011; 52: 1867-72.
22. Pfreundschuh M, Ho AD, Cavallin-
Stahl E, et al. Prognostic significance of
maximum tumour (bulk) diameter in
young patients with good-prognosis diffuse
large-B-cell lymphoma treated with
CHOP-like chemotherapy with or without
rituximab: an exploratory analysis of the
MabThera International Trial Group (MInT)
study. Lancet Oncol 2008; 9: 435-44.
23. Lowry L, Smith P, Qian W, et al. Reduced
dose radiotherapy for local control
in non-Hodgkin lymphoma: a randomised
phase III trial. Radiother Oncol 2011; 100:
86-92.
24. Miller TP. Management of intermediate-
grade lymphomas. Oncology (Williston
Park) 1998; 12: Suppl 8: 35-9.
25. Gordon KB, Char DH, Sagerman RH.
Late effects of radiation on the eye and
ocular adnexa. Int J Radiat Oncol Biol
Phys 1995; 31: 1123-39.
Copyright © 2015 Massachusetts Medical Society.