Un hombre de 21 años ingresó en este hospital debido a artralgias, disnea e infiltrados pulmonares.
El paciente había estado bien hasta 14 meses antes del ingreso, cuando desarrolló una inflamación del párpado inferior izquierdo. Vio a su médico quien le prescribió olopatadina y eritromicina oftálmica. Once meses antes del ingreso, desarrolló hinchazón y eritema de ambos párpados inferiores e hinchazón dolorosa entre el primero y segundo metacarpiano derechos. Se le prescribió ibuprofeno y cetirizina. Dos meses después, desarrolló dolor torácico pleurítico y disnea, y se auscultó un frote pericárdico; se realizó un diagnóstico de pericarditis. El paciente fue tratado con ibuprofeno y los síntomas se resolvieron.
Un mes más tarde, el paciente vio a su médico debido a fatiga persistente, mialgias, dolor de rodilla y sudores nocturnos. Impresionaba enfermo; el pulso era de 120 a 130 latidos por minuto, y el resto del examen fue normal. Los índices de glóbulos rojos, el recuento de plaquetas y el nivel de tirotropina fueron normales, y las pruebas para detectar la IgG del virus de Epstein-Barr y anticuerpos heterófilos fueron negativos; otros resultados de la prueba se muestran en la Tabla 1



Tabla 1 Datos de laboratorio.
Una radiografía de tórax reveló una masa. La tomografía computarizada (TC) del tórax, realizada después de la administración de material de contraste, reveló opacidades pulmonares parcheadas multifocales y un quiste, de 1 cm de diámetro, en el ápice izquierdo. Se prescribieron levofloxacina, suplementos de hierro y multivitaminas. Durante las siguientes 3 semanas, se desarrollaron malestar general, congestión nasal, conjuntivitis, prurito generalizado y empeoramiento de la fatiga. Se prescribieron antihistamínicos, glucocorticoides nasales y ketotifeno fumarato solución oftálmica. Siete meses antes de esta admisión, había presentado fiebre intermitente hasta una temperatura de 38.6 ° C que se asociaron con escalofríos, disnea e hinchazón de la mano derecha; Los síntomas no mejoraron con ibuprofeno. El paciente fue ingresado al hospital.
Al ingreso, el paciente refirió disnea por la mañana y con el esfuerzo; congestión nasal, con mucosidad clara; mialgias difusas; erupciones pruriginosas en sus mejillas; aumento de la sed y delvolumen de diuresis; pérdida de peso involuntaria de 3,6 kg durante un período de 2 meses; y dolores en las articulaciones que afectaban el cuello, los hombros, las caderas, los tobillos y las rodillas, dolores que eran peores en la mañana. No reportó tos, dolor de espalda ni disuria. Tenía antecedentes de acné y rinitis alérgica. Los medicamentos incluían peróxido de benzoilo y ácido retinoico. Tenía una alergia a la penicilina (que se manifestaba con erupción). Había nacido en el noreste de los Estados Unidos de padres que habían emigrado de Haití. Era un estudiante universitario y vivía en un dormitorio. No fumaba, no tomaba alcohol ni consumía drogas ilícitas, y no era sexualmente activo. Su abuela materna tenía artritis reumatoide, y su tía había fallecido a los 58 años de edad de lupus eritematoso sistémico (LES).
En el examen, la temperatura era de 37,7 ° C; había dolor en los tendones del bíceps y ganglios linfáticos palpables (iguales o menores de 1 cm), en las regiones cervical posterior, axilar e inguinal; el resto del examen era normal. Los niveles en sangre de electrolitos, glucosa, bilirrubina total y directa y proteína total fueron normales, al igual que las pruebas de función renal; las pruebas de detección de factor reumatoide, anticuerpos IgG contra péptidos citrulinados (CCP), anticuerpos anti-La (SSB) y virus de la hepatitis B y C fueron negativos. Otros resultados de las pruebas se muestran en la Tabla 1. Una radiografía de tórax fue normal. Un ecocardiograma transtorácico mostró una función ventricular derecha e izquierda normales, una fracción de eyección estimada del 64%, un pequeño derrame pericárdico (sin evidencia de taponamiento) y una presión sistólica estimada del ventrículo derecho de 27 mmHg. El análisis de orina reveló trazas de proteinuria. Los cultivos de sangre y orina eran estériles; el examen de las heces no mostró protozoos ni toxina de Clostridium difficile. El paciente fue dado de alta dentro de las 24 horas posteriores al ingreso, con instrucciones para tomar ibuprofeno, hierro, vitaminas y difenhidramina y usar un inhalador nasal de budesonida. Se le diagnosticó una posible enfermedad de Still de inicio en la edad adulta.
Durante las siguientes 2 semanas, se desarrollaron artralgias de la mano derecha. Las pruebas de detección de una mutación V617F (1849G → T) en el gen de la quinasa 2 de Janus (JAK2) y de autoanticuerpos anti-U1-ribonucleoproteína (U1-RNP) fueron negativas; El nivel de glucosa-6-fosfato deshidrogenasa fue normal. Otros resultados de las pruebas se muestran en la Tabla 1. Se prescribieron ibuprofeno y prednisona, 15 mg diarios. Durante el mes siguiente, se desarrolló una erupción que fue intermitentemente pruriginosa y se produjeron episodios de hinchazón de las manos. En el examen, la piel estaba seca, con máculas y pápulas hiperpigmentadas de 2 a 5 mm de diámetro en las nalgas, la espalda y los brazos; Algunos tenían costras y la mayoría eran foliculares. El examen patológico de una muestra de biopsia de piel mostró foliculitis aguda.
Un mes antes de esta admisión, las fiebres aumentaron en frecuencia, con tos y artralgias recurrentes. En la evaluación, la temperatura era de 39,2 ° C, había pápulas acneiformes en la espalda y había una leve linfadenopatía axilar. Una radiografía de tórax era normal. Los niveles de complemento fueron normales, y la prueba de anticuerpos contra el ADN de doble cadena y La (SSB) fue negativa; otros resultados de la prueba se muestran en la Tabla 1. Se prescribió naproxeno.
Cuatro semanas más tarde, en una visita de seguimiento, el paciente informó alguna mejoría en las fiebres, pero aumento de la tos y la disnea en el esfuerzo, la hinchazón periorbitaria, el eritema, y el edema de las manos y los dedos. El examen reveló edema periorbitario y eritema bilaterales, ganglios linfáticos palpables en el lado derecho del cuello (1,5 cm de diámetro) y axila izquierda (2 cm de diámetro), ganglios linfáticos inguinales, una erupción acneiforme en el tronco y los hombros con lesiones hiperpigmentadas postinflamatorias, hinchazón dolorosa de las pequeñas articulaciones de las manos y pequeños derrames en ambas rodillas; El resto del examen era normal. Los niveles en sangre de electrolitos, glucosa, calcio, fósforo, magnesio, bilirrubina total y directa, proteína total, fosfatasa alcalina, ácido láctico, péptido natriurético N-terminal pro-tipo B, enzima convertidora de angiotensina y proteína inhibidora C1 fueron normales, como lo fueron las pruebas de función tiroidea y renal y el tiempo parcial de tromboplastina; otros resultados de la prueba se muestran en la Tabla 1. Más tarde ese día, los síntomas respiratorios empeoraron y el paciente acudió al servicio de urgencias. El pulso era de 102 latidos por minuto, otros signos vitales eran normales y el examen físico no había cambiado. Una radiografía de tórax mostró una opacidad irregular en la base del pulmón izquierdo. Fue ingresado nuevamente en el hospital.
Las pruebas serológicas para anticuerpos contra el virus de inmunodeficiencia humana (VIH) tipos 1 y 2, bartonella, proteinasa 3, mieloperoxidasa, Jo-1, anticuerpos antineutrófilos citoplasmáticos y anticuerpos antimitocondriales fueron negativos, como pruebas para anticuerpos contra proteinasa 3, mieloperoxidasa y Jo-1; Otros resultados de las pruebas estaban pendientes. Un cultivo nasofaríngeo fue negativo. La TC del tórax reveló opacidades parcheadas, bilaterales, multifocales, subpleurales, reticulares y en vidrio esmerilado en los pulmones, más en los lóbulos inferiores que en los lóbulos superiores.
La tomografía computarizada del tórax realizada sin la administración de material de contraste reveló opacidades reticulares subpleurales bilaterales y en vidrio esmerilado, con una afectación más grave del lóbulo inferior izquierdo (Figura). Había pérdida de volumen en el lóbulo inferior izquierdo, con desplazamiento posterior de la cisura mayor izquierda en comparación con la cisura mayor derecha. Una reconstrucción coronal mostró falta de afinamiento distal de los bronquiolos en el lóbulo inferior izquierdo, lo que es consistente con la bronquiolectasia por tracción. La combinación de opacidades reticulares subpleurales, pérdida de volumen y bronquiolectasias por tracción sugiere un proceso intersticial fibrótico. No había evidencia de panalización. No había masas pulmonares, linfadenopatías mediastínicas ni derrames pleurales.
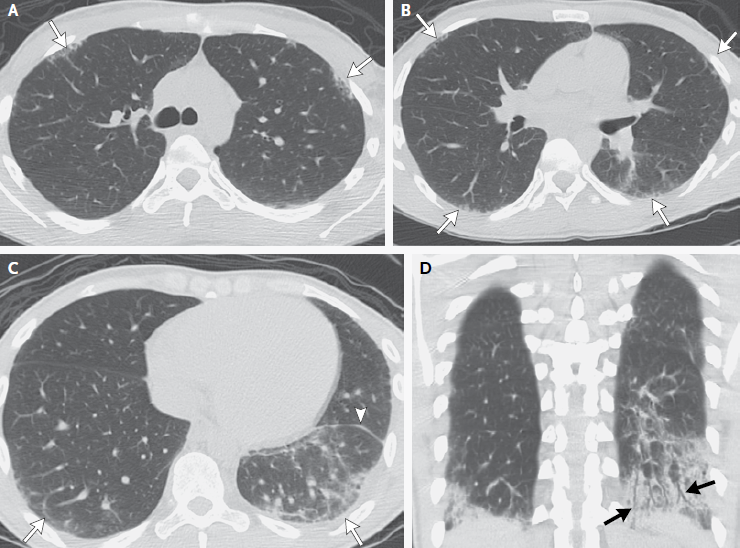
Figura 1. Imágenes de TC del tórax de lainternación.
Las imágenes de TC axial seleccionadas del tórax superior (Panel A), medio (Panel B) e inferior (Panel C) muestran opacidades reticulares subpleurales bilaterales y en vidrio esmerilado (flechas), con la mayor afectación del lóbulo inferior izquierdo. El desplazamiento posterior de la cisura mayor izquierda (Panel C, punta de flecha) sugiere pérdida de volumen en el lóbulo inferior izquierdo. Una reconstrucción coronal (Panel D) muestra falta de afinamiento de los bronquiolos distales en el lóbulo inferior izquierdo (flechas), una característica consistente con la bronquiolectasia de tracción. Estos hallazgos sugieren un proceso intersticial fibrótico.
Se realizaron pruebas de diagnóstico adicionales y un procedimiento de diagnóstico.
DIAGNÓSTICO DIFERENCIAL
Este hombre previamente sano de 21 años con fiebre, artritis, erupción cutánea, lesiones pulmonares y antecedentes familiares de enfermedad autoinmune tuvo una enfermedad prolongada que comenzó 14 meses antes de su segundo ingreso en este hospital.
El diagnóstico diferencial consta de tres categorías: infección, cáncer y enfermedad vascular del colágeno. Discutiré brevemente posibles diagnósticos en las primeras dos categorías, y luego me concentraré en las enfermedades colágeno vasculares, la causa más probable de la enfermedad de este hombre. Las características sobresalientes de este caso que ayudan a enfocar nuestro diagnóstico diferencial son erupción con prurito en las mejillas del paciente, hinchazón bilateral de los párpados y eritema, una historia de serositis (pericarditis), tos, linfadenopatía, síntomas constitucionales, fiebres episódicas que aumentan en frecuencia, involuntariamente pérdida de peso, sudores nocturnos, artritis, mialgias y, quizás lo más importante para ayudar a reducir nuestro diagnóstico diferencial, infiltrados pulmonares.
ENFERMEDAD PULMONAR INTERSTICIAL
El diagnóstico diferencial de las opacidades en vidrio esmerilado es amplio. En este caso, los hallazgos de la TC coinciden con la enfermedad pulmonar intersticial. La enfermedad pulmonar intersticial puede ser idiopática o asociada con una causa identificable. Los subtipos pueden distinguirse sobre una base clínica o patológica. La fibrosis pulmonar idiopática es el tipo más común. Otros incluyen neumonitis intersticial aguda, neumonía organizativa criptogénica (anteriormente conocida como bronquiolitis obliterante con neumonía en organización), neumonía intersticial descamativa, neumonía intersticial linfocítica, bronquiolitis respiratoria asociada a enfermedad pulmonar intersticial y neumonitis intersticial inespecífica. Este paciente tiene una enfermedad pulmonar intersticial, pero la pregunta es si es una forma idiopática o si está asociada con un proceso de enfermedad específico.
INFECCIÓN
En este paciente con síntomas constitucionales, linfadenopatía y erupción, la infección por VIH es una consideración. La enfermedad pulmonar intersticial puede desarrollarse en pacientes con el síndrome de inmunodeficiencia adquirida, más comúnmente como resultado de infecciones oportunistas, pero también se han observado cánceres y neumonitis intersticial linfocítica1,2. La prueba de VIH de este paciente fue negativa.
Los síntomas del paciente también provocan la consideración de bartonella, toxoplasmosis, mononucleosis infecciosa, histoplasmosis, parvovirus y sífilis. Los pacientes con cualquiera de estas infecciones pueden presentar erupción cutánea, fiebre, linfadenopatía, artritis o una combinación de estas, pero la enfermedad pulmonar intersticial no es una característica importante de ninguna de ellas. En este paciente, las pruebas serológicas para bartonella y mononucleosis fueron negativas. La sífilis es poco probable si el paciente no es sexualmente activo. Otras enfermedades virales, como coxsackievirus o adenovirus, pueden haber tenido un papel en la enfermedad inicial, pero no explican la eventual constelación de síntomas.
CÁNCER
La mielofibrosis idiopática crónica, un trastorno mieloproliferativo, es un diagnóstico posible. La mielofibrosis debe considerarse cuando un recuento de glóbulos blancos notablemente elevado no tiene otra causa obvia, a pesar de un diagnóstico. Este diagnóstico se consideró en este caso, ya que el estudio incluyó pruebas para la mutación 1849G → T (V617F) en JAK2, que fue negativa. Esta mutación somática es un marcador molecular de policitemia vera (en el 95% de los casos), trombocitosis esencial y mielofibrosis idiopática (en aproximadamente el 50% de los casos). Un resultado positivo de la prueba indica una neoplasia mieloide clonal, pero un resultado negativo no descarta tales neoplasias. Aunque la mielofibrosis idiopática sigue siendo una posibilidad en este caso, la enfermedad pulmonar intersticial asociada, la erupción cutánea y la artritis justifican una mayor exploración.
ENFERMEDADES COLÁGENO VASCULARES
Una enfermedad colágeno vascular es muy probable en este paciente, dada la fuerte historia familiar, la constelación de síntomas y los hallazgos de laboratorio. Los diagnósticos más probables son la enfermedad de Still del adulto, el LES, la artritis reumatoide, el síndrome de Sjögren, la enfermedad mixta del tejido conectivo y la dermatomiositis (en el contexto del síndrome antisintetasa).
ENFERMEDAD DE STILL DEL ADULTO
Este paciente recibió un diagnóstico provisional de enfermedad de Still de inicio en la edad adulta durante su primer ingreso en este hospital. Hasta la fecha, se han propuesto ocho conjuntos de criterios diagnósticos para la enfermedad de Still de aparición en la adultez, y casi todos incluyen las características principales de fiebre, leucocitosis, erupción cutánea, artritis y artralgia; sin embargo, los criterios de Yamaguchi tienen la mayor sensibilidad.3,4 De acuerdo con los criterios de Yamaguchi, que consisten en criterios mayores y menores, el diagnóstico de la enfermedad de Still de aparición en adultos requiere al menos cinco características, incluidos al menos dos criterios principales.
Este paciente tenía fiebre, artritis y leucocitosis. Su erupción fue inconsistente con la erupción de color salmón notable en la enfermedad de Still de adulto; sin embargo, la erupción en pacientes con tonos oscuros de piel puede aparecer hiperpigmentada en lugar de color salmón. También tuvo linfadenopatía, resultados anormales de las pruebas de función hepática y pruebas negativas para anticuerpos antinucleares (FAN) y factor reumatoide. Los niveles elevados de ferritina, aunque no están incluidos en los criterios de diagnóstico, suelen estar presentes en pacientes con enfermedad de Still de inicio en adultos. En este caso, la prueba del nivel de ferritina no se realizó inicialmente, pero luego se encontró que el nivel era elevado. Un nivel elevado de ferritina es un hallazgo inespecífico que probablemente represente un reactante de fase aguda en el contexto de una enfermedad inflamatoria.
En ausencia de otra explicación, la enfermedad de Still de inicio en adultos continuaría siendo alta en el diagnóstico diferencial. Sin embargo, la enfermedad pulmonar intersticial se reporta raramente en combinación con la enfermedad de Still de inicio en la edad adulta. 5
LUPUS ERITEMATOSO SISTÉMICO
Las características del LES en este paciente incluyen artritis, serositis, erupción malar y anticuerpos anti-Ro (SSA). La linfadenopatía se puede ver en LES.5,6 Tanto la infección como la actividad de la enfermedad son causas comunes de fiebre en pacientes con LES. La enfermedad pulmonar intersticial rara vez se asocia con LES.7 Una prueba de FAN negativa (como se señala en este paciente) tiene una sensibilidad del 99% para descartar LES.
ARTRITIS REUMATOIDE
En general, la fiebre no es típica de los pacientes con artritis reumatoide en ausencia de infección, y la enfermedad pulmonar intersticial, aunque a menudo está presente, suele ser asintomática. La prevalencia de enfermedad pulmonar intersticial en pacientes con artritis reumatoide está entre el 5% y el 10%, y los pacientes con ambos trastornos tienen un mal pronóstico. 8-10 La linfadenopatía no es una característica típica de la artritis reumatoide, pero puede ocurrir; el riesgo de linfoma entre los pacientes con artritis reumatoide es mayor que el riesgo en la población en general. No se observa una erupción, excepto en casos de superposición con otras enfermedades vasculares del colágeno (por ejemplo, LES y dermatomiositis). La ausencia de factor reumatoide y autoanticuerpos anti-CCP tendería a descartar la artritis reumatoide.
SÍNDROME DE SJOGREN
Los autoanticuerpos anti-Ro, un sello del síndrome de Sjögren, estuvieron presentes en este paciente en dos ocasiones. La enfermedad pulmonar intersticial es la anomalía pulmonar más común en el síndrome de Sjögren primario.11 La enfermedad pulmonar intersticial, la fiebre y la artritis pueden estar asociadas con el síndrome de Sjögren, pero en este caso no se presentaron síntomas sicca, otra característica de esta enfermedad.
ENFERMEDAD MIXTA DE TEJIDO CONECTIVO
Aunque existe cierto debate sobre si se trata de una entidad de diagnóstico independiente, 12 la enfermedad mixta del tejido conectivo tiene características de miositis, LES y esclerosis sistémica. También hay criterios serológicos. Los pacientes con enfermedad mixta del tejido conectivo deben tener características clínicas (edema de las manos, sinovitis, miositis, fenómeno de Raynaud y acrosclerosis) y la presencia de autoanticuerpos anti-U1-RNP. Este paciente no tenía U1-RNP, lo que nos permite descartar este diagnóstico.
DERMATOMIOSITIS
Creo que la dermatomiositis, en el contexto del síndrome antisintetasa, es el diagnóstico más probable. La polimiositis y la dermatomiositis se clasifican según los criterios desarrollados en 1975 por Bohan y Peter.13 La naturaleza sistémica de estas enfermedades, incluidas las características distintas de la participación de los músculos y la piel, se ha ido reconociendo cada vez más en los últimos 30 años.
El síndrome antisintetasa, definido hace dos décadas, 14,15 se caracteriza por la presencia de uno de los autoanticuerpos antisintetasa y al menos una de las siguientes tres características clínicas: enfermedad pulmonar intersticial, miopatía inflamatoria o poliartritis inflamatoria (Tabla 2). Otras características que apoyan el diagnóstico de síndrome antisintetasa pueden incluir fiebre, fenómeno de Raynaud y "manos mecánicas" (piel gruesa y agrietada, generalmente en las palmas y las superficies radiales de los dedos). 20


Tabla 2 Anticuerpos específicos de la miositis.
Hasta la fecha, se han identificado ocho autoanticuerpos antisintetasa, todos los cuales están dirigidos contra enzimas que acetilan ARN de transferencia (ARNt). El primero descrito y más comúnmente encontrado es el anti-Jo-1 (dirigido contra la histidil-ARNt sintetasa). Aunque son menos comunes, los otros siete autoanticuerpos de este grupo (anti-PL-12, anti-PL-7, anti-EJ, anti-OJ, anti-KS, anti-YRS y anti-Zo) confieren una similar fenotipo clínico.21 La mayoría de los estudios han demostrado que la mayoría de los pacientes con autoanticuerpos antisintetasa tienen enfermedad pulmonar intersticial.22 En contraste, la proporción de pacientes con miositis y enfermedad pulmonar intersticial que tienen autoanticuerpos antisintetasa conocidos se estima entre un tercio y dos tercios.23 No se sabe cuántos pacientes con enfermedad pulmonar intersticial tienen autoanticuerpos anti-Jo-1 u otros autoanticuerpos antisintetasa en la presentación. Un estudio retrospectivo informó pruebas serológicas para detectar la presencia de autoanticuerpos antisintetasa en 198 pacientes con neumonías intersticiales24. Se encontraron autoanticuerpos antisintetasa en 13 de los pacientes (6,6%), ampliando nuestra comprensión de las neumonías intersticiales que se creía que eran idiopáticas. Se han descrito otras antisintetasas, particularmente anti-PL-12 (dirigidas contra alanil-ARNt sintetasa) en pacientes con enfermedad pulmonar intersticial en ausencia de miositis clínicamente aparente, lo que puede llevar a confusión y retraso en el diagnóstico. Aunque la mayoría de los anticuerpos antisintetasa pueden analizarse comercialmente, solo el Jo-1 puede detectarse rutinariamente mediante un ensayo inmunoenzimático. Los otros requieren pruebas especializadas en laboratorios de referencia. En este paciente, no se detectaron autoanticuerpos anti-Jo-1.
PISTAS PARA EL DIAGNÓSTICO
El edema del párpado y el eritema con prurito son muy característicos de la erupción en heliotropo asociada con la dermatomiositis. El eritema malar también ocurre y difiere de la típica "erupción en alas de mariposa" del LES. En el LES, los pliegues nasolabiales son respetados por la erupción, mientras que en la dermatomiositis, no lo son. Otras características sobresalientes del síndrome de antisintetasa en este paciente incluyen enfermedad pulmonar intersticial, artritis, fiebre, niveles marcadamente elevados de enzimas musculares (creatina quinasa, aldolasa y las aminotransferasas, con el nivel de aspartato aminotransferasa más alto que el nivel de alanina aminotransferasa), y quizás lo más importante, una prueba de FAN negativa pero una prueba positiva para autoanticuerpos anticitoplásmicos.
LA IMPORTANCIA DE LOS AUTOANTICUERPOS ANTICITOPLÁSMICOS
Ocasionalmente, cuando se ordena una prueba de FAN, se observa una tinción anticitoplasmática en su lugar. Los anticuerpos anticitoplasmáticos reaccionan con estructuras subcelulares visualmente reconocibles (mitocondrias, aparato de Golgi o citoesqueleto) o antígenos citoplásmicos, incluidos Ro, anticuerpos antimitocondriales, partículas de reconocimiento de señal y sintetasas de ARNt.26 En este paciente, la mayoría de los anticuerpos anticitoplasmáticos en mayores títulos son más probablemente autoanticuerpos antisintetasa no–Jo-1 tales como anti–PL-7 o anti–PL-12.
ANTICUERPOS ANTI-RO
El Ro de 60 kD es un autoantígeno nuclear, mientras que el Ro de 52 kD es citoplásmico y se observa a menudo en concierto con los autoanticuerpos antisintetasa. La presencia de estos dos anticuerpos anti-Ro junto con los autoanticuerpos antisintetasa puede conferir riesgo de enfermedad pulmonar más grave27.
Dada la constelación de signos y síntomas de este paciente en presencia de posibles autoanticuerpos antisintetasa, mi diagnóstico es dermatomiositis en el contexto del síndrome de antisintetasa (muy probablemente con autoanticuerpos asociados con anti-PL-12 o anti-PL-7). La prueba de diagnóstico más útil sería un panel de autoanticuerpos de miositis para evaluar los autoanticuerpos antisintetasa y una biopsia de pulmón a cielo abierto para confirmar la enfermedad pulmonar intersticial.
La sangre se envió a un laboratorio de referencia para analizar los anticuerpos asociados a la miositis y se realizó una biopsia de pulmón con toracoscopia asistida por video.
DIAGNOSTICO CLÍNICO PRESUNTIVO
DERMATOMIOSITIS EN EL CONTEXTO DEL SÍNDROME ANTISINTETASA (MUY PROBABLEMENTE CON AUTOANTICUERPOS ANTI-PL-12 O ANTI-PL-7).
DISCUSION PATOLOGICA
Se realizó una biopsia toracoscópica de pulmón. El pulmón estaba firme en la palpación en la cirugía, y la muestra estaba firme en el examen general. El examen de los cortes reveló un patrón mixto. Inflamación granulomatosa que involucraba bronquiolos (Figura 2A) y fibrosis mixoideorganizativa en bronquiolos respiratorios, un elemento de la bronquiolitis obliterante (Figura 2B), estaban presentes. Otras regiones mostraron neumonía organizada crónica con fibrosis y simplificación de los espacios aéreos (Figura 2C) y fibrosis intersticial más difusa con inflamación (Figura 2D) en un patrón que podría describirse como una neumonitis intersticial no específica. Algunos bronquiolos estaban llenos de neutrófilos y contenían purulencia, características de una bronquiolitis aguda. También había pleuritis crónica con fibrosis pleural.
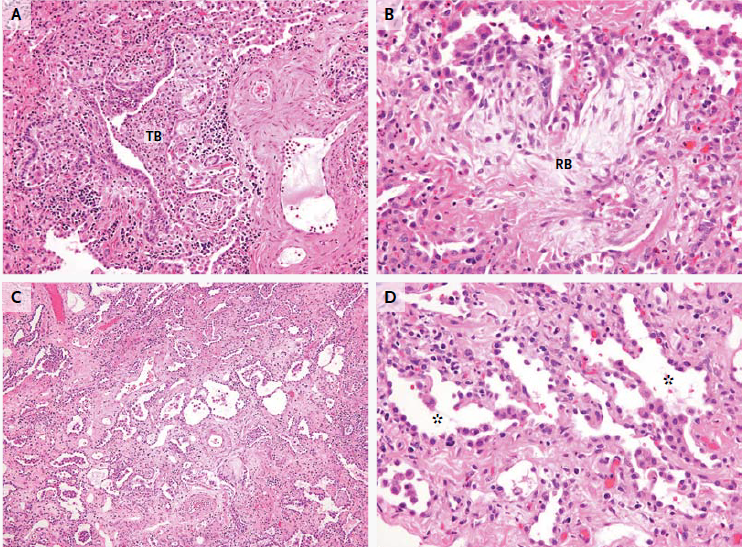

Figura 2 Muestra de la biopsia de pulmón.
El panel A (hematoxilina y eosina) muestra una inflamación linfocítica e histiocítica que llena y obstruye un bronquiolo terminal más grande (TB). El panel B (hematoxilina y eosina) muestra bronquiolitis obliterante, con tejido fibroso mixoide azul que obstruye un bronquiolo respiratorio (RB) más pequeño. El panel C (hematoxilina y eosina) muestra fibrosis fija con simplificación de los espacios aéreos debajo de la pleura. Algunas áreas muestran fibrosis intersticial difusa (Panel D, hematoxilina y eosina), con neumocitos hipertróficos que recubren los alvéolos (asteriscos). Una arteria elástica tiene capas elásticas internas y externas y la luz (Panel E, flechas; tinción elástica) notablemente comprometida por la proliferación de la íntima (asterisco).
Cuando el patólogo no puede clasificar una neumonitis intersticial como una entidad definida única, como la neumonitis intersticial habitual, la neumonitis intersticial inespecífica o la neumonía organizada criptogénica, un patrón mixto puede sugerir enfermedad vascular del colágeno, reacción fármacos o aspiración, entre otras afecciones. La variación en los patrones puede deberse en parte a la fase de la enfermedad. En la enfermedad vascular del colágeno, la enfermedad pulmonar difiere según la enfermedad subyacente. Por ejemplo, los nódulos necrotizantes pueden ocurrir en la artritis reumatoide, la capilaritis en el lupus eritematoso y en la esclerodermia puede desarrollarse un marcado cambio hipertensivo pulmonar. Los patrones de neumonitis intersticial inespecífica y neumonía organizada criptogénica son comunes a todas las enfermedades del tejido conectivo, incluida la dermatomiositis. Ninguno de los hallazgos en este caso nos permitió considerar seriamente una enfermedad del tejido conectivo sobre otra.
Otra forma de abordar el estado de este paciente es considerar la neumonitis asociada a miositis. Aunque los informes clínicos y radiográficos de enfermedad pulmonar asociada con el síndrome anti-PL-12 son numerosos, 25,28-35 hay poco en la literatura que ilustre las características histopatológicas. Se ha dicho que los pacientes tienen neumonitis intersticial inespecífica, fibrosis pulmonar idiopática o neumonitis intersticial aguda, pero estos informes generalmente no incluyen los resultados de la biopsia. La neumonitis intersticial se ha ilustrado en algunos casos. Los hallazgos clínicos, radiológicos e histopatológicos en este paciente con miositis se explican mejor como parte del síndrome de antisintetasa.
La bronquiolitis aguda puede ser una infección superpuesta, pero no se puede descartar como parte integral aguda del proceso. También hubo un cambio de hipertensión arterial pulmonar (Figura 2E), que es común en pacientes con cicatrización de los pulmones, pero se ha descrito en algunos otros pacientes con síndrome de antisintetasa36, 37
DISCUSIÓN DEL MANEJO
Finalmente, el diagnóstico del síndrome antisintetasa se estableció en este caso con una prueba positiva para anticuerpos anti-PL-12. En casi todos los pacientes con el síndrome antisintetasa anti-PL-12, se desarrolla una enfermedad pulmonar; los pacientes pueden presentar enfermedad pulmonar, como lo hizo este paciente. Es posible que la miositis clínicamente significativa nunca se desarrolle: este paciente tenía mialgias y niveles elevados de creatina quinasa, pero no tenía debilidad documentada.
El pronóstico para este paciente es el de un paciente con una enfermedad crónica.28 En una serie de síndrome antisintetasa la tasa de supervivencia a 10 años fue del 70% para los pacientes con autoanticuerpos anti-Jo-1 y del 47% para los pacientes con otros autoanticuerpos antisintetasa; ya que este paciente cae en la última categoría, su pronóstico, desafortunadamente, es malo. La causa más común de muerte en la serie fue la fibrosis pulmonar progresiva y la hipertensión pulmonar. Este paciente ya tiene evidencia de hipertensión pulmonar por la muestra de la biopsia.
No conocemos ensayos controlados sobre el tratamiento del síndrome antisintetasa que nos guíe en la selección de la terapia para este paciente. Los agentes inmunosupresores, típicamente glucocorticoides, con metotrexato o azatioprina como los agentes adicionales más comunes, se usan típicamente y pueden resultar en una mejoría clínica en pacientes con miositis inflamatoria. Se ha demostrado que la gamma globulina intravenosa es eficaz en un estudio controlado de dermatomiositis.38
Este paciente continuó teniendo enfermedad pulmonar progresiva, fiebre, miositis y artritis y requirió dosis variables de ibuprofeno, prednisona y metotrexato para controlar sus síntomas. La erupción típica del síndrome de antisintetasa (manos del mecánico) apareció en el curso de la enfermedad (Figura 3) y, por lo tanto, el paciente ha tenido todas las características de este síndrome. Fue capaz de continuar sus cursos universitarios de forma intermitente durante 2 años después del diagnóstico, a pesar de esta grave enfermedad crónica.

Figura 3. Fotografía clínica de la mano del paciente
La TC de tórax de seguimiento realizada 21 meses después del ingreso reveló un aumento en las opacidades reticulares subpleurales y el desarrollo de múltiples espacios quísticos subpleurales, que probablemente representan un proceso de panalización precoz. Estos cambios son consistentes con la progresión de la enfermedad pulmonar intersticial (Figura4).

Figura 4: Imágenes de tomografía computarizada de seguimiento obtenidas 21 meses después de la admisión.
Los paneles (superior), B (medio), y C (inferior) de tórax muestran un aumento de las opacidades reticulares subpleurales con desarrollo de múltiples espacios quísticos subpleurales, hallazgo consistente con panalización (flechas). Una reconstrucción coronal (panel D) muestra leves bronquiectasias de tracción (flecha). Estos hallazgos son consistentes con progresión d la enfermedad pulmonar intersticial.
Desafortunadamente, dos años y medio después de esta admisión, se desarrollaron nuevos problemas, como meningitis aséptica, convulsiones, neumotórax recurrente y pericarditis complicada por taquicardia ventricular y paro cardíaco. Después del paro cardíaco, el paciente presentó hipotensión prolongada y desarrolló fiebre, leucopenia y síndrome de activación macrofágica. Fue tratado con anakinra (un antagonista de la interleucina-1), y los aspectos inflamatorios de su enfermedad mejoraron, pero sigue teniendo una recuperación lenta. Exigió amputaciones transmetatarsianas bilaterales debido a necrosis isquémica, una complicación de la hipotensión prolongada y el tratamiento vasopresor del shock.
DIAGNOSTICO ANATOMICO
ENFERMEDAD PULMONAR INTERSTICIAL (BRONQUIOLITIS Y NEUMONITIS INTERSTICIAL INESPECÍFICA) Y MIOSITIS CON AUTOANTICUERPOS ANTI-PL-12 (SÍNDROME DE ANTISINTETASA).
Traducción de :
“A 21-Year-Old Man with Fevers, Arthralgias, and Pulmonary Infiltrates”
Lisa Christopher-Stine, M.D., M.P.H., Dwight R. Robinson, M.D., Carol C. Wu, M.D., and Eugene J. Mark, M.D.
N Engl J Med 2012; 367:2134-2146November 29, 2012DOI: 10.1056/NEJMcpc1208147
References
1
de Leon FC, Britt EJ. The noninfectious respiratory complications of infection with HIV. Curr Opin Pulm Med 1995;1:223-233
.
2
Grieco MH, Chinoy-Acharya P. Lymphocytic interstitial pneumonia associated with the acquired immune deficiency syndrome. Am Rev Respir Dis 1985;131:952-955
Web of Science
.
3
Masson C, Le Loet X, Liote F, et al. Comparative study of 6 types of criteria in adult Still's disease. J Rheumatol 1996;23:495-497
Web of Science | Medline
.
4
Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol 1992;19:424-430
Web of Science | Medline
.
5
Ohta A, Yamaguchi M, Tsunematsu T, et al. Adult Still's disease: a multicenter survey of Japanese patients. J Rheumatol 1990;17:1058-1063
Web of Science | Medline
.
6
Shapira Y, Weinberger A, Wysenbeek AJ. Lymphadenopathy in systemic lupus erythematosus: prevalence and relation to disease manifestations. Clin Rheumatol 1996;15:335-338
CrossRef | Web of Science | Medline
.
7
Eisenberg H, Dubois EL, Sherwin RP, Balchum OJ. Diffuse interstitial lung disease in systemic lupus erythematosus. Ann Intern Med 1973;79:37-45
Web of Science | Medline
.
8
Koduri G, Norton S, Young A, et al. Interstitial lung disease has a poor prognosis in rheumatoid arthritis: results from an inception cohort. Rheumatology (Oxford) 2010;49:1483-1489
CrossRef | Web of Science
.
9
Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006;39:233-241
CrossRef | Web of Science
.
10
Young A, Koduri G. Extra-articular manifestations and complications of rheumatoid arthritis. Best Pract Res Clin Rheumatol 2007;21:907-927
CrossRef | Web of Science
.
11
Cain HC, Noble PW, Matthay RA. Pulmonary manifestations of Sjögren's syndrome. Clin Chest Med 1998;19:687-699
CrossRef | Web of Science
.
12
Zimmermann C, Steiner G, Skriner K, Hassfeld W, Petera P, Smolen JS. The concurrence of rheumatoid arthritis and limited systemic sclerosis: clinical and serologic characteristics of an overlap syndrome. Arthritis Rheum 1998;41:1938-1945
CrossRef | Web of Science
.
13
Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med 1975;292:344-347
Full Text | Web of Science | Medline
.
14
Marguerie C, Bunn CC, Beynon HL, et al. Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. Q J Med 1990;77:1019-1038
CrossRef | Web of Science | Medline
.
15
Marie I, Hatron PY, Hachulla E, Wallaert B, Michon-Pasturel U, Devulder B. Pulmonary involvement in polymyositis and in dermatomyositis. J Rheumatol 1998;25:1336-1343
Web of Science
.
16
Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol 2011;65:25-34
CrossRef | Web of Science
.
17
Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011;63:713-721
CrossRef | Web of Science
.
18
Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford) 2009;48:607-612
CrossRef | Web of Science
.
19
Betteridge ZE, Gunawardena H, McHugh NJ. Novel autoantibodies and clinical phenotypes in adult and juvenile myositis. Arthritis Res Ther 2011;13:209-209
CrossRef | Web of Science
.
20
Connors GR, Christopher-Stine L, Oddis CV, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years? Chest 2010;138:1464-1474
CrossRef | Web of Science
.
21
Mimori T, Imura Y, Nakashima R, Yoshifuji H. Autoantibodies in idiopathic inflammatory myopathy: an update on clinical and pathophysiological significance. Curr Opin Rheumatol 2007;19:523-529
CrossRef | Web of Science
.
22
Targoff IN. Myositis specific autoantibodies. Curr Rheumatol Rep 2006;8:196-203
CrossRef
.
23
Targoff IN. Autoantibodies and their significance in myositis. Curr Rheumatol Rep 2008;10:333-340
CrossRef
.
24
Watanabe K, Handa T, Tanizawa K, et al. Detection of antisynthetase syndrome in patients with idiopathic interstitial pneumonias. Respir Med 2011;105:1238-1247
CrossRef | Web of Science
.
25
Kalluri M, Sahn SA, Oddis CV, et al. Clinical profile of anti-PL-12 autoantibody: cohort study and review of the literature. Chest 2009;135:1550-1556
CrossRef | Web of Science
.
26
Koh WH, Dunphy J, Whyte J, Dixey J, McHugh NJ. Characterisation of anticytoplasmic antibodies and their clinical associations. Ann Rheum Dis 1995;54:269-273
CrossRef | Web of Science
.
27
La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006;39:249-253
CrossRef | Web of Science
.
28
Cassidy EA, Aggarwal R, Fertig N, Lucas M, Ascherman DP, Oddis CV. Non-Jo-1 anti-synthetase autoantibodies are not specific for myositis and are associated with poor survival. Arthritis Rheum 2010;62:Suppl:929-929
CrossRef | Web of Science
.
29
Fischer A, Swigris JJ, du Bois RM, et al. Anti-synthetase syndrome in ANA and anti-Jo-1 negative patients presenting with idiopathic interstitial pneumonia. Respir Med 2009;103:1719-1724
CrossRef | Web of Science
.
30
Hirakata M, Suwa A, Nagai S, et al. Anti-KS: identification of autoantibodies to asparaginyl-transfer RNA synthetase associated with interstitial lung disease. J Immunol 1999;162:2315-2320
Web of Science
.
31
Hoagland MB, Stephenson ML, Scott JF, Hecht LI, Zamecnik PC. A soluble ribonucleic acid intermediate in protein synthesis. J Biol Chem 1958;231:241-257
Web of Science | Medline
.
32
Mehta P, Patel L, Roncaroli F, Pickering M, Brand A. Painful myositis in the antisynthetase syndrome with anti-PL12 antibodies. J Transl Med 2010;8:8-8
CrossRef | Web of Science
.
33
Satoh S, Hirakata M, Suwa A, Mimori T, Inada S, Akizuki M. Two cases of interstitial pneumonia with anti-PL-12 (alanyl tRNA synthetase) antibodies. Ryumachi 1996;36:862-868
.
34
Vega P, Ibarra M, Prestridge A, Pachman LM. Autoantibody to PL-12 (anti-ananyl-tRNA synthetase) in an African American girl with juvenile dermatomyositis and resolution of interstitial lung disease. J Rheumatol 2011;38:394-395
CrossRef | Web of Science
.
35
Yahaba M, Suda A, Syoji R, Jujo T, Shinozaki T. A case of rapidly progressive interstitial pneumonia with anti-pL-12-antibodies successfully treated by pulse methylprednisolone and cyclophosphamide. Nihon Kokyuki Gakkai Zasshi 2008;46:547-551
.
36
Handa T, Nagai S, Kawabata D, et al. Long-term clinical course of a patient with anti PL-12 antibody accompanied by interstitial pneumonia and severe pulmonary hypertension. Intern Med 2005;44:319-325
CrossRef | Web of Science
.
37
Hervier B, Wallaert B, Hachulla E, et al. Clinical manifestations of anti-synthetase syndrome positive for anti-alanyl-tRNA synthetase (anti-PL12) antibodies: a retrospective study of 17 cases. Rheumatology (Oxford) 2010;49:972-976
CrossRef | Web of Science
.
38
The Muscle Study Group. A randomized, pilot trial of etanercept in dermatomyositis. Ann Neurol 2011;70:427-436
CrossRef | Web of Science