![]() |
| Hospital Municipal "Dr Ángel Pintos" |
Mujer de 32 años
MOTIVO DE INTERNACIÓN: extrasistolia ventricular con criterios de malignidad
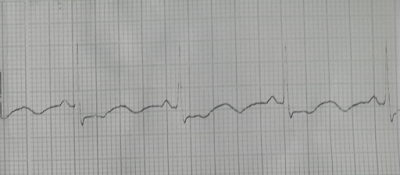
ENFERMEDAD ACTUAL: es traída a la guardia del hospital por servicio de emergencia por haberse despertado con “sensación de muerte”, malestar general, mareos. Inicialmente interpretada como “crisis nerviosa” se aplicó diacepan 10mg IM y se realiza ECG que muestra aritmia compleja por extrasístoles ventriculares frecuentes que ceden espontáneamente al igual que todo el cuadro.
ANTECEDENTES DE ENFERMEDAD ACTUAL: antecedentes de episodios de nerviosismo, episodios fugaces de palpitaciones. Diarrea que atibuye a “estados nerviosos”.Quince días antes de la internación actual consultó a su médico por palpitaciones por lo que se reduce la dosis de T4 que toma desde hace varios años. Posteriormente se reciben dosajes de hormonas tiroideas solicitados al momento de dicha consulta (antes de modificar la dosis): TSH 0,41mU/L (normal 0,4 a 4,5). T4 libre 2,2ng/dl (normal 0,7 a 1,8).
Dos días antes de esta internación recibió analgésicos por contractura cérvico-dorsal. Refiere asimismo en los últimos 7 días calambres en gemelos.
ANTECEDENTES PERSONALES Y PATOLÓGICOS: Hipotiroidismo desde hace 13 años. HTA diagnosticada hace 1 año. Le explicaron que la misma se relacionaba con su "personalidad nerviosa". "Extrasistolia", refiere que le han diagnosticado extrasístoles en varias consultas y algunos electrcardiogramas pero que siempre le dijeron que eran "benignas" y que no requerían estudio ni tratamiento específico. Según le dijo su madre tuvo sufrimiento fetal al final de su gesta.
Fecha de última menstruación 1 mes antes. Eva test actual (-) Tomó anticonceptivos hasta hace 1 mes
ANTECEDENTES FAMILIARES: Madre arritmia. Padre: ACV, DBT2, HTA, Dislipemia
Abuelo fallecido de muerte súbita a los 50 años
HÁBITOS: alcohol escaso los fines de semana, tabaco no. Dieta general. Medicación actual valsartan 80 mg atenolol 50 mg y 25 mg de hidroclorotiazida/día, levotiroxina 125 ug/día
Trabaja como empleada en un comercio
EXAMEN FÍSICO: lúcida, TA 170/100 mm Hg frecuencia 65 por minuto. Temperatura 36,2°C
Frecuencia respiratoria 20 por minuto. Saturación de O2 98% respirando aire ambiente.
Tórax y abdomen s/p
Se solicitan análisis de rutina y de los cuales llama la atención Hipopotasemia severa 1,1 meq/l, Na 147, creatinina 2 mg/dl. y CPK 7905 UI/L
Ionograma en orina: Na: 54,2 K: 27,25, Cl: 61,1. Volumen diurético 2600 ml/24 hs
Se indica potasio EV, Mg y K por vía oral.
Interconsulta con cardiología: Paciente con HTA tratada crónicamente con valsartán, diuréticos y betabloqueantes que ingresa por arritmia ventricular asociada a QT largo, HVI. Se objetiva hipopotasemia severa. Pendiente eco 2D. Se interpreta como hipopotasemia asociada a diuréticos.
Informe del ECG: Ritmo sinusal. FC 71 Eje eléctrico de QRS 0* Onda P. QTc 566 mes PR 140 mseg QRS 90 mseg. Infradesnivel horizontal del ST en dos derivaciones que creo son DI, DII, AVF. Sin embargo no tanto en DIII ni en AVL. Pero sí es infradesnivel de 2 mm y horizontal que traduce lesión subendocardica. Arritmia ventricular extra sistolica con tripletas, dupletas polimorficas. Ecocardiograma y eco Doppler no muestran HVI, FS conservada, pericardio libre, resto s/p. Se solicitó ecografía abdominal y TC para evaluar suprarrenales.
![]() |
D1
|
![]() |
D2
|
![]() |
D3
|
![]() |
| aVR |
![]() |
| aVL aVF |
![]() |
V1 V2 V3
|
![]() |
| V4 |
![]() |
| V5 V6 |
![]() |
| RITMO |
Ecocardiograma y eco Doppler no muestran HVI, FS conservada, pericardio libre, resto s/p.Se solicitó ecografía abdominal y TC para evaluar suprarrenales.
Tomografía computada de abdomen: litiasis vesicular. Se observa imagen hipodensa de 27 X 17 mm en suprarrenal derecha que realza con la administración de contraste EV compatible con adenoma.
![]() |
| La flecha marca el adenoma adrenal derecho |
![]() |
| Añadir leyenda |
Después de tomar muestras de sangre para aldosterona, renina, cortisol plasmático, CLU, ácido vanil mandélico y catecolaminas plasmáticas se comenza con espironolactona 200 mg/día, potasio por VO, y eventual enalapril si la TA es mayor a 140/90 mm Hg.
Se reciben los valores de laboratorio hormonales:
Actividad de renina plasmática: menos de 0,6 ng/mL/hora.
Aldosterona plasmática: 30 ng/dl
Relación Aldosterona/renina 50.
Valores de CLU, vanilmandélico y catecolaminas plasmáticas y urinarias dentro de los valores normales
Se interpreta el cuadro como hiperaldosteronismo primario secundario a adenoma suprarrenal derecho funcionante. Se consideró innecesario el dosaje diferencial de aldosterona en ambas venas suprarrenales para confirmar lateralización y se indicó cirugía laparoscópica de la glándula adrenal derecha a lo que la paciente y su esposo se opusieron. A la paciente se le ofreció continuar con tratamiento médico con espironolactona 100mg cada 12 horas y controles clínicos e imagenológicos por consultorio externo.
Seis meses y un año después del diagnóstico, la paciente se encuentra asintomática, su TA es de 128/86 mmHg,supotasio plasmático es de 4,1 meq/L y el tamaño del adenoma no se ha modificado.
HIPERALDOSTERONISMO PRIMARIO (SÍNTESIS)
Aunque inicialmente se consideró una rareza, el aldosteronismo primario ahora se considera una de las causas más comunes de hipertensión secundaria ( HTA ). Litynski informó los primeros casos, pero Conn fue el primero en caracterizar bien el trastorno en 1956. El síndrome de Conn, como se describió originalmente, se refiere específicamente al aldosteronismo primario debido a la presencia de un aldosteronoma suprarrenal (neoplasia suprarrenal benigna secretora de aldosterona).
Con base en datos más antiguos, originalmente se estimó que el aldosteronismo primario representaba menos del 1% de todos los pacientes con HTA. Datos posteriores, sin embargo, indicaron que en realidad puede ocurrir hasta en un 5-15% de los pacientes con HTA. Aldosteronismo primario puede ocurrir en un porcentaje aún mayor de pacientes con HTA resistente al tratamiento y puede estar considerablemente infradiagnosticado; esto es especialmente cierto si los pacientes con HTA refractaria al tratamiento no son remitidos específicamente para su evaluación a un endocrinólogo.
Aunque el aldosteronismo primario sigue siendo un desafío diagnóstico considerable, el reconocimiento de la enfermedad es crítico porque la HTA asociada al aldosteronismo primario a menudo se puede curar (o al menos controlar de forma óptima) con la intervención quirúrgica o médica adecuada. El diagnóstico generalmente es de 3 niveles, lo que implica un cribado inicial, una confirmación del diagnóstico y una determinación del subtipo específico de aldosteronismo primario.
Aunque estudios previos sugirieron que los aldosteronomas eran la causa más común de aldosteronismo primario (70-80% de los casos), el trabajo epidemiológico posterior indicó que la prevalencia de aldosteronismo debido a hiperplasia suprarrenal idiopática bilateral (HIB), es mayor de lo que había sido creído previamente. Estos informes sugieren que la HIB puede ser responsable de hasta el 75% de los casos de aldosteronismo primario. Además, los informes han descrito un raro síndrome de aldosteronismo primario caracterizado por características histológicas intermedias entre el adenoma suprarrenal y la hiperplasia suprarrenal, que a menudo se localiza unilateralmente (también se refirió a la literatura anterior como "aldosteronismo intermedio") .
Figura 1. Imagen de resonancia magnética (MRI) en un paciente con síndrome de Conn que muestra un adenoma suprarrenal izquierdo.
Figura 2. El escintigrama obtenido mediante el uso de yodo-131-beta-yodometil-norcolesterol (NP-59) en un hombre de 59 años con hipertensión muestra una captación de radionúclidos bastante intensa en el tumor adrenal derecho. En la cirugía, se confirmó un tumor de Conn.
Clínicamente, la distinción entre las 2 principales causas del aldosteronismo primario es vital porque el tratamiento de elección para cada uno es marcadamente diferente. Si bien el tratamiento de elección para los aldosteronomas es la extirpación quirúrgica, el tratamiento de elección para la HIB es el tratamiento médico con antagonistas de la aldosterona.
Las entidades que se sabe que causan aldosteronismo incluyen lo siguiente:
- Adenomas productores de aldosterona (APA) [ 1 ]
- Adenomas productores de aldosterona sensible a la renina (AP-RAs, también abreviado como RRAs)
- Hiperplasia suprarrenal idiopática bilateral (glomerulosa) o HIB (también conocida como hiperplasia suprarrenal primaria o HSP)
- Formas familiares de aldosteronismo primario
- Secreción ectópica de aldosterona (los ovarios y los riñones son los dos órganos descritos en la literatura que, en el contexto de una enfermedad neoplásica, que pueden ser fuentes ectópicas de aldosterona, pero esto es raro).
- Carcinomas suprarrenales productores de aldosterona puros (muy raros; fisiológicamente se comportan como APA)
La aldosterona, al inducir la reabsorción renal de sodio en el túbulo contorneado distal (TCD), aumenta la secreción de iones de potasio y de hidrógeno, causando hipernatremia, hipocalemia y alcalosis.
ALDOSTERONISMO PRIMARIO GENÉTICO-FAMILIAR
Existen tres variedades genético-familiares distintas de aldosteronismo primario. Sutherland y sus colegas describieron por primera vez la variedad tipo 1 de aldosteronismo primario familiar, aldosteronismo remediable con glucocorticoides (GRA), en 1966. En GRA, la HTA responde clínicamente a pequeñas dosis de glucocorticoides además de otros agentes antihipertensivos. [ 2 ]La forma tipo 1 de aldosteronismo primario familiar se debe a un producto génico quimérico aberrante formado que combina el promotor sensible a glucocorticoides (inhibidor) del gen de la 11beta-hidroxilasa (CYP11B1) con la región codificante del gen de la aldosterona sintetasa (CYP11B2). En niveles de glucocorticoides ambientales, el promotor no se silencia por completo de la transcripción, y esto conduce a la sobreexpresión de la aldosterona sintetasa, con el consiguiente aumento de la síntesis y secreción de aldosterona.
La variante tipo 2 del aldosteronismo primario familiar (que no es sensible a los glucocorticoides) se describió por primera vez en 1991. Aunque no se ha identificado la anomalía genética exacta para el aldosteronismo primario tipo 2, los datos sugieren que el locus para esta enfermedad está en la banda 7p22. [ 3 ]
La variante tipo 3 del aldosteronismo primario familiar se debe a KCNJ5 (canal de rectificación interior del potasio, subfamilia J, miembro 5) mutaciones del canal de potasio. Este tipo fue descrito por el grupo Lifton en 2011. [ 4 ]
DIAGNÓSTICO
1 PRUEBAS DE DETECCIÓN (DE PRIMER NIVEL) PARA EL ALDOSTERONISMO PRIMARIO INCLUYEN LAS SIGUIENTES:
ANiveles séricos de potasio y bicarbonato
B Niveles de sodio y magnesio
C Relación de actividad plasmática de aldosterona / renina plasmática
2 LAS PRUEBAS DE CONFIRMACIÓN (SEGUNDO NIVEL) INCLUYEN LO SIGUIENTE:
A Nivel de aldosterona sérica
B Prueba de excreción de aldosterona en orina de 24 horas
C Prueba de carga de sal
3 PRUEBAS PARA DETERMINAR EL SUBTIPO DE ALDOSTERONISMO PRIMARIO (PRUEBAS DE TERCER NIVEL) INCLUYEN LAS SIGUIENTES:
A Prueba de estimulación postural
B Prueba de estimulación de furosemida (Lasix)
C Ritmo diurno de la aldosterona
LA INVESTIGACIÓN RADIOLÓGICA INICIAL EN EL ESTUDIO DEL ALDOSTERONISMO PRIMARIO ES LA TOMOGRAFÍA COMPUTARIZADA (TC) SUPRARRENAL DE ALTA RESOLUCIÓN Y CORTE FINO (2-2,5 mm) CON CONTRASTE.
Otras pruebas incluyen lo siguiente:
CENTELLOGRAFÍA CON YODO-METIL-NORCOLESTEROL NP-59: aunque es bastante difícil de establecer y no está disponible de forma rutinaria, esta prueba puede ser útil en casos seleccionados para distinguir entre adenomas e hiperplasia.
MUESTREO VENOSO SUPRARRENAL: el muestreo venoso suprarrenal probablemente tiene su mayor utilidad cuando los hallazgos de imágenes suprarrenales son completamente normales a pesar de la evidencia bioquímica de aldosteronismo primario y en escenarios en los que la patología suprarrenal bilateral está presente en imágenes y la bioquímica sugiere la presencia de un aldosteronoma funcional
PRUEBA DE SUPRESIÓN DE DEXAMETASONA: esta prueba es relevante solo en el contexto de un posible aldosteronismo familiar
PRUEBA DE METOCLOPRAMIDA (REGLAN): esta es una prueba no invasiva para distinguir entre aldosteronomas e hiperplasia suprarrenal idiopática (HSI)
MANEJO
La terapia farmacológica incluye el uso de lo siguiente:
- Bloqueadores de los canales de calcio
- Antagonistas de mineralocorticoides
- Glucocorticoides
La cirugía es el tratamiento de elección para las variantes lateralizables del aldosteronismo primario, incluidos los aldosteronomas típicos, los adenomas sensibles a la renina (RRA) y la hiperplasia suprarrenal primaria (HAP). Una adrenalectomía se puede realizar a través de una laparotomía formal o mediante el uso de una técnica laparoscópica (con la utilización de este último método cada vez más común).
FISIOPATOLOGÍA
- Los factores más importantes que aseguran la hipocalemia en el aldosteronismo primario son: hipersecreción de aldosterona, que actúa sobre el conducto colector cortical para estimular la secreción de potasio en el líquido tubular, aumentando así la pérdida renal / urinaria de potasio [ 5 ] ;
- Volumen intravascular adecuado, que permite un suministro adecuado de agua (flujo tubular) a los túbulos contorneados distales (TCD) y los conductos colectores para permitir la pérdida renal de potasio;
- Ingesta adecuada de sodio en la dieta, lo que a su vez aumenta el potasio corporal total, el suministro renal / tubular de sodio y, por lo tanto, aumenta la pérdida renal de potasio a través del sistema de transporte en contracorriente.
La ausencia de una o más de las circunstancias fisiológicas descritas anteriormente puede explicar la ausencia de hipocalemia franca en muchos pacientes con aldosteronismo primario comprobado.
La alcalosis metabólica asociada en el aldosteronismo primario se debe a una mayor pérdida renal de iones de hidrógeno mediada por hipocalemia y aldosterona.
Casi el 20% de los pacientes con aldosteronismo primario tienen intolerancia a la glucosa como resultado del efecto inhibidor de la hipocalemia sobre la acción y secreción de la insulina; sin embargo, la diabetes mellitus no es más común que en la población general.
ETIOLOGÍA
La anomalía cardinal que causa el síndrome de aldosteronismo primario es la producción de aldosterona autónoma (no suprimible). Además de la producción de aldosterona no suprimible, los niveles de renina plasmática suprimidos coexisten conun volumen volumen de líquido intratravascular y extravascular muy levemente expandido. La regulación normal de la secreción de aldosterona está mediada en diversos grados por la renina, los niveles séricos de potasio y sodio, el estado del volumen intravascular y la corticotropina.
La regulación de la producción de aldosterona por estos factores puede alterarse de diversas maneras, según el subtipo de aldosteronismo primario. Generalmente, los adenomas productores de aldosterona (APA) y GRA siguen siendo sensibles a la corticotropina, mientras que la hiperplasia suprarrenal idiopática (IAH) y los adenomas sensibles a la renina (RRA) productores de aldosterona mantienen la respuesta al sistema renina-angiotensina (RAS).
En aldosteronismo remediable con glucocorticoides (GRA), se suprime el RAS y la aldosterona se regula mediante corticotropina debido a la fusión del gen quimérico de un promotor sensible a la corticotropina con las regiones codificantes del gen de la aldosterona sintetasa (que normalmente no tiene dicho promotor ) Por lo tanto, los niveles de corticotropina ambiental patológicamente sobreestimulan la síntesis de aldosterona inapropiadamente. [ 6 ]
En pacientes con aldosteronismo remediable con glucocorticoides (GRA), la administración de dexametasona (o cualquier otro glucocorticoide) a dosis suficientes para suprimir la producción excesiva de corticotropina da como resultado una reducción en la síntesis de aldosterona y natriuresis y la eventual corrección de las anomalías bioquímicas del aldosteronismo primario. [ 7 ] Los estudios histológicos en esta enfermedad han mostrado hiperplasia específica de la zona fasciculada, con atrofia concomitante de la zona glomerular.
CAUSAS
La causa exacta del aldosteronismo primario esporádico debido a un adenoma o hiperplasia no está clara. La existencia de factores tróficos (p. Ej., Endotelinas, citocinas) se ha postulado en casos de hiperplasia. Las mutaciones somáticas de los genes que conducen a una ventaja de crecimiento en el tejido adenomatoso suprarrenal son una causa posible, pero no probada.
En formas familiares de aldosteronismo primario, se conoce la base molecular de GRA. GRA se debe a una mutación que resulta de un producto genético híbrido. [ 3 ] Los genes de la 11beta-hidroxilasa y la aldosterona sintetasa que normalmente se encuentran cerca uno del otro en el cromosoma 8 se cruzan para crear un nuevo producto genético híbrido. Este gen híbrido consiste en la secuencia reguladora sensible a la corticotropina del gen de 11beta-hidroxilasa (CYP11B1) fusionado al componente estructural del gen de la aldosterona sintetasa (CYP11B2). [ 2 ]
La mayoría de los aldosteronomas esporádicos surgen de la zona fasciculada, y a menudo tienen una hiperplasia glandular cercana al adenoma. Esto sugiere que se produce una respuesta proliferativa de las células a algún factor paracrino / autocrino actualmente no identificado. Dentro de esta zona de hiperplasia, se cree que tiene lugar un cambio clonal en una sola célula, proporcionando así el nido para el adenoma en desarrollo.
La base genética del aldosteronismo familiar tipo 2 no está clara; sin embargo, el locus para esta enfermedad se ha mapeado en 7p22 (banda 11q13). [ 3 ] Este síndrome puede manifestarse histológicamente como hiperplasia o adenomas.
La base genética para el aldosteronismo familiar tipo 3 se ha descifrado recientemente. Las mutaciones en el gen codificador del canal de potasio KCNJ5 dan como resultado la pérdida de selectividad iónica, la despolarización de la membrana celular, el aumento de la entrada de Ca2 + en las células adrenales de la glomerulosa y el aumento de la síntesis de aldosterona. [ 4 ]
ALDOSTERONISMO TERCIARIO
La existencia de aldosteronismo terciario como una entidad separada sigue siendo controvertida. Se presume que la entidad es el resultado de elevaciones crónicas de los niveles de renina en plasma y aldosteronismo secundario, lo que finalmente establece un estado de aldosteronismo no regulado y autónomo con una imagen histológica de hiperplasia mixta y adenomas en el tejido adrenocortical afectado. Se considera que este cuadro clinicopatológico es el resultado final irreversible de los efectos neurohumorales prolongados sobre la resistencia vascular y la hipertrofia "terminal" del tejido adrenocortical productor de aldosterona.
Existen pocos casos bien descritos, pero en la mayoría, las glándulas suprarrenales son hiperplásicas, a menudo con hiperplasia nodular (que puede causar confusión diagnóstica). Prácticamente todos los casos descritos se encuentran en el contexto de la estenosis de la arteria renal, lo que complica aún más la atribución del estado hipertensivo al exceso crónico de aldosterona "inapropiada".
Inicialmente, los niveles de renina son elevados, lo que es típico del aldosteronismo secundario. Cuando se desarrolla la fase terciaria (autónoma), el perfil bioquímico cambia a un estado de renina baja / alta aldosterona. El paradigma es análogo a la patogénesis del hiperparatiroidismo terciario.
EPIDEMIOLOGÍA
PREVALENCIA EN LOS ESTADOS UNIDOS
La prevalencia exacta del aldosteronismo primario no está clara, pero las estimaciones sugieren que el 5-15% de los casos de hipertensión esencial (HTA) pueden deberse al aldosteronismo primario. La prevalencia de aldosteronismo primario es probablemente más alta en pacientes que tienen un nivel de potasio sérico bajo, en pacientes que son ancianos y en pacientes que tienen HTA que es resistente al uso de medicamentos únicos.
PREVALENCIA INTERNACIONAL
No hay evidencia que demuestre que el aldosteronismo primario, en sus formas más comunes, ocurra en exceso relativo en cualquier parte del mundo. [ 8 ]
DATOS DEMOGRÁFICOS RELACIONADOS CON LA RAZA, EL SEXO Y LA EDAD
El aldosteronismo primario ocurre en todo el mundo. Varios informes sugieren una mayor prevalencia entre los afroamericanos, las personas de origen africano y, potencialmente, otros negros. Esto parece ser particularmente cierto de la variante idiopática de hiperplasia suprarrenal (IAH) de la enfermedad.
Los adenomas productores de aldosterona (APA) son más comunes en mujeres que en hombres, con una relación de mujer a hombre de 2: 1. El paciente típico con APA es una mujer de entre 30 y 50 años.
PRONÓSTICO
La morbilidad y la mortalidad asociadas con el aldosteronismo primario, especialmente el síndrome de Conn, se relacionan principalmente con la hipocalemia y la hipertensión (HTA). [ 9 , 10 ] La hipocalemia, especialmente si es grave, causa arritmias cardíacas, que pueden ser fatales.
Las complicaciones de la HTA crónica incluyen infarto de miocardio, enfermedad cerebrovascular e insuficiencia cardíaca congestiva. El tratamiento también puede conducir a complicaciones, como reacciones a los medicamentos y complicaciones de la cirugía.
Existe evidencia de que el aldosteronismo crónico en sí mismo, en ausencia de presión arterial elevada (p. Ej., Como ocurre en el aldosteronismo secundario), también se asocia con un mayor riesgo de lesión cardíaca, que incluye lesión isquémica, hipertrófica y fibrótica. Además, los estudios han demostrado que los pacientes con aldosteronismo primario son más propensos a tener o desarrollar hipertrofia ventricular izquierda, accidente cerebrovascular y síndromes coronarios agudos que los pacientes con grados similares de HTA por otras causas. [ 11 ] Por supuesto, los pacientes con HTA por aldosteronismo primario también corren el riesgo de desarrollar todo el espectro de complicaciones de la HTA crónica, incluida la nefropatía hipertensiva y la retinopatía.
HISTORIA
La presentación clínica del aldosteronismo primario no es distintiva, y el diagnóstico correcto requiere un alto índice de sospecha por parte del médico. Los escenarios clínicos comunes en los que se debe considerar la posibilidad de aldosteronismo primario incluyen los siguientes:
Pacientes con hipocalemia espontánea o no provocada, especialmente si el paciente también es hipertenso [ 12 ]
Pacientes que desarrollan hipocalemia severa y / o persistente en el contexto de dosis bajas a moderadas de diuréticos que deplecionan de potasio
Pacientes con hipertensión resistente al tratamiento (HTA refractaria)
Los pacientes con hipocalemia severa reportan fatiga, debilidad muscular, calambres, dolores de cabeza y palpitaciones. También pueden tener polidipsia y poliuria por diabetes insípida nefrogénica inducida por hipocalemia. La HTA de larga duración puede provocar problemas cardíacos, retinianos, renales y neurológicos, con todos los síntomas y signos asociados.
En un estudio que comparó los efectos cardíacos del aldosteronismo primario versus secundario, Cesari et al determinaron que si bien ambos tipos de aldosteronismo se caracterizaban con frecuencia por hipertrofia ventricular izquierda y disfunción diastólica, solo el aldosteronismo primario se asoció con una evidente disfunción sistólica subclínica. Además, los pacientes con aldosteronismo primario tuvieron frecuencias cardíacas más bajas y valores de resistencia arterial y resistencia vascular más altos que aquellos con la condición secundaria, mientras que la actividad de la renina plasmática fue menor en el aldosteronismo primario que en el secundario (0,56 frente a 15,00 ng / ml / h, respectivamente). . [ 13 ]
ALDOSTERONISMO PRIMARIO FAMILIAR
Las dos principales variedades familiares de aldosteronismo primario son el aldosteronismo remediable por glucocorticoides (GRA, aldosteronismo primario familiar tipo 1) y un tipo no remediable por glucocorticoides (aldosteronismo primario familiar tipo 2).
El reconocimiento de GRA es particularmente importante debido a sus implicaciones para los pacientes que son hipertensos y cuyos miembros de la familia aparentemente no se ven afectados. La HTA, derrames cerebrales y otros eventos cardiovasculares significativos se describen en personas jóvenes con este síndrome.
Aunque el síndrome es poco común, los niveles elevados de sospecha son esenciales para el diagnóstico. Menos de 200 casos bien validados existen en la literatura. Todos los pacientes con GRA deben ser tratados médicamente con glucocorticoides y sin cirugía.
Aunque poco común, GRA puede ser más frecuente de lo que se suponía previamente. Un subgrupo significativo de pacientes con la variedad normopotasémica más leve de este síndrome probablemente se presume incorrectamente que tiene una HTA esencial. [ 9 ]
Los antecedentes familiares de HTA (particularmente con una edad temprana de inicio), HTA en niños, HTA de baja renina y presunta HIA son las situaciones típicas en las que se debe considerar este diagnóstico.
El tercer tipo de PA familiar, debido a mutaciones en el gen codificador del canal de potasio KCNJ5, se considera extremadamente raro, pero también puede conducir a HTA e hipocalemia a una edad muy temprana.
EXAMEN FÍSICO
Los pacientes con aldosteronismo primario no presentan hallazgos clínicos distintivos, y un alto índice de sospecha basado en la historia del paciente es vital para realizar el diagnóstico. Los hallazgos podrían incluir lo siguiente:
Hipertensión (HTA) - Esta condición casi invariablemente ocurre, aunque se han descrito en la literatura algunos casos raros de aldosteronismo primario no asociado con HTA.
Debilidad
Distensión abdominal
Íleo por hipokalemia
Hallazgos relacionados con complicaciones de la HTA: insuficiencia cardíaca, hemiparesia debida a accidente cerebrovascular, soplos carotídeos, soplos abdominales, proteinuria, insuficiencia renal, encefalopatía hipertensiva (confusión, cefalea, convulsiones, cambios en el nivel de conciencia) y cambios retinianos hipertensivos
Es importante tener en cuenta que el aldosteronismo primario en sí mismo por lo general no se asocia con edema, a pesar del estado de volumen expandido asociado con este. La falta de edema es el resultado de natriuresis espontánea y diuresis (llamado "escape de aldosterona") que ocurre en pacientes con aldosteronismo primario y que parece estar mediado por el péptido natriurético auricular (ANP). [ 14 , 15 ] Cabe señalar que este efecto probablemente se basa en la activación del sistema apical ATP / UTP / P2Y2 (en el nivel del túbulo de conexión / conducto colector), lo que conduce a una mayor presentación de sodio, que a su vez induce cierre del canal de sodio epitelial (ENaC), con la disminución resultante en la reabsorción de sodio (es decir, natriuresis potenciada). [ 16] Por lo tanto, el hallazgo de edema importante en los pacientes que se presume que tienen aldosteronismo sugiere que o bien un diagnóstico equivocado o se ha hecho que las complicaciones asociadas, como la insuficiencia renal o cardiaca, están presentes.
CONSIDERACIONES DIAGNÓSTICAS
Se debe considerar el diagnóstico de aldosteronismo primario en todas las personas con hipertensión (HTA) e hipocalemia. Hacer el diagnóstico correcto puede ser la única forma de lograr un control adecuado de la presión arterial y, por lo tanto, prevenir las secuelas de una HTA mal controlada.
Las condiciones a considerar en el diagnóstico diferencial del aldosteronismo primario incluyen las siguientes:
HTA
HTA maligna
Encefalopatía hipertensiva
Hipocalemia
Alcalosis metabólica
Estenosis de la arteria renal
HTA Renovascular
HTA esencial con baja renina: Constituye aproximadamente el 40% de la HTA esencial
Masticar tabaco
Intoxicación por carbenoxolona
Síndrome aparente de exceso de mineralocorticoides (AME)
Varias causas de aldosteronismo secundario: a diferencia del aldosteronismo primario, estas causas se asocian con niveles elevados de renina (actividad de renina plasmática)
Síndrome de Chrétien: este raro síndrome se caracteriza por un exceso de mineralocorticoides y una HTA adrenocortical secundaria a un adenoma pituitario que produce proopiomelanocortina (POMC) [ 17 ]
Tumor suprarrenal secretor de deoxycorticosterone (DOC)
Isquemia renovascular
Preeclampsia (toxemia del embarazo)
Tumor secretor de renina: estos son tumores raros que surgen del aparato yuxtaglomerular
Consumo excesivo de regaliz: en esta situación, el componente de ácido glicirricínico inhibe la 11beta-hidroxiesteroide deshidrogenasa, lo que altera la conversión de cortisol en cortisona en los riñones; por lo tanto, el cortisol se une a los receptores mineralocorticoides y actúa como un mineralocorticoide
Otros trastornos y síndromes genéticos / familiares seleccionados a considerar incluyen los siguientes:
Síndrome de Gitelman - Esto se debe a un cotransportador de sodio / cloruro defectuoso (NCCT); es básicamente una tubulopatía perdedora de sal con aldosteronismo secundario
Síndrome de Barrter: es una fenocopia de al menos 3 defectos genéticos distintos (es decir, hiperactividad del cotransportador de sodio-dicloruro de potasio [NKCC2], el canal de potasio medular externo renal [ROMK1] o el canal de cloruro epitelial renal [ClCKb], este último codificado por el gen barttin, también es una tubulopatía que pierde sal con aldosteronismo secundario y es fisiopatológicamente similar al síndrome de Gitelman
Síndrome de Gordon: esto se debe a mutaciones inactivantes de las serina-treonina quinasas WNK1 y WNK4 ("sin lisina [K]" quinasas), lo que provoca hipertensión, hipercalemia, hipercloremia leve, acidosis y actividad suprimida de la renina plasmática
Pseudoaldosteronismo (síndrome de Liddle): este es un trastorno autosómico dominante raro debido a mutaciones hiperactivantes del canal de sodio epitelial renal (ENaC), con una reabsorción excesiva de sodio en el túbulo distal renal; los niveles de renina y aldosterona son bajos
Deficiencia de 11beta-hidroxiesteroide deshidrogenasa
Resistencia a glucocorticoides: esto se debe a mutaciones inactivantes del receptor de glucocorticoides
DIAGNÓSTICOS DIFERENCIALES
Carcinoma suprarrenal
Incidentaloma suprarrenal
Cirugía suprarrenal
Síndrome de Bartter
Deficiencia de hidroxilasa C-11
Deficiencia de hidroxilasa C-17
Complejo de Carney
Síndrome de Conn
Eclampsia
Síndrome de Cushing Iatrogénico
CONSIDERACIONES DIAGNÓSTICAS
Los individuos con aldosteronismo primario pueden presentar alcalosis metabólica hipopotasémica; sin embargo, hasta el 38% de los pacientes con aldosteronismo primario pueden ser normocalémicos al momento de la presentación. [ 12 ]
Los estudios de laboratorio de rutina pueden mostrar hipernatremia, hipocalemia y alcalosis metabólica como resultado de la acción de la aldosterona en el túbulo contorneado distal renal (mejora la reabsorción de sodio y la excreción de iones de potasio y de hidrógeno). [ 18 ]
PRUEBAS HISTÓRICAS
FLEBOGRAFÍA SUPRARRENAL
Este procedimiento se intentó en la década de 1980 y su objetivo era visualizar de forma invasiva los patrones venosos que rodean a los adenomas adrenocorticales. El procedimiento ha caído en descrédito debido al riesgo de infarto suprarrenal y ya no se usa.
Ensayo terapéutico de espironolactona (Aldactone)
Este procedimiento ya no se usa como una prueba de diagnóstico para aldosteronismo primario, porque existen alternativas más fáciles y más rápidas; por lo tanto, actualmente tiene un valor histórico.
Por incompleto, el ensayo terapéutico con espironolactona involucró la administración de espironolactona por vía oral a una dosis de 100 mg 4 veces al día durante 5 semanas ya no se usa. Una prueba positiva se caracterizaría por una disminución en la presión arterial diastólica de al menos 20 mm Hg.
PRUEBAS DE DETECCIÓN (PRIMER NIVEL)
NIVELES SÉRICOS DE POTASIO Y BICARBONATO
La hipopotasemia y la alcalosis metabólica tienen bajas sensibilidades y especificidades para el aldosteronismo primario cuando estos niveles se prueban por sí mismos. La hipopotasemia ( nivel sérico de potasio menor de 3.6 mEq / l) tiene una sensibilidad del 75-80% mientras el paciente sigue una dieta normal de sodio. [ 5 ] Por lo general, se asocia con alcalosis metabólica leve (niveles séricos de bicarbonato mayores de 31 mEq / l) y una kaliuresis inadecuada (excreción urinaria de potasio mayor de 30 mmol / día).
NIVELES DE SODIO Y MAGNESIO
La hipernatremia sérica leve en el rango de 143-147 mEq / L y la hipomagnesemia leve por pérdida renal de magnesio son otros hallazgos bioquímicos asociados en el aldosteronismo primario establecido. [ 19 ]
RELACIÓN DE ACTIVIDAD PLASMÁTICA DE ALDOSTERONA / RENINA PLASMÁTICA
Dado a que la relación aleatoria de la actividad plasmática de aldosterona / renina plasmática (PRA) es bastante constante en muchas condiciones fisiológicas, puede utilizarse para el cribado. Los valores normales son menores a 270 cuando la concentración de aldosterona se expresa en pmol / L, o son menores de 10 cuando la concentración de aldosterona se expresa en ng / dL. [ 20 ]
ALGORITMO 1. PARA LA DETECCIÓN DE POTENCIAL ALDOSTERONISMO PRIMARIO.
Cuando la aldosterona se mide en ng / dL y la ARP se mide en ng / ml / h, una relación de aldosterona en plasma / ARP superior a 20-25 tiene una sensibilidad del 95% y una especificidad del 75% para el aldosteronismo primario. Cuando la aldosterona se mide en pmol / l, una relación mayor a 900 es consistente con el aldosteronismo primario. [ 21 , 22 ]
LIMITACIONES
Las limitaciones en la utilidad de la ración de aldosterona / ARP en plasma incluyen lo siguiente:
Una de las principales limitaciones de estas pruebas es la variabilidad inherente de la secreción de aldosterona debido a un ritmo circadiano intrínseco
La mayoría de las recomendaciones sugieren realizar la prueba mientras se retiran todos los antihipertensivos que pueden afectar el sistema renina-angiotensina (RAS); esto puede ser difícil de lograr cuando una enfermedad grave dicta la continuación de algunos medicamentos para controlar la hipertensión (HTA) y la hipopotasemia durante la prueba.
La obtención de la relación aldosterona / ARP en plasma en el contexto del uso crónico de un inhibidor de la enzima convertidora de angiotensina (ACE) (es decir, más de 4 semanas de uso) aumenta la especificidad de la prueba de relación pero reduce la sensibilidad
Aunque la utilidad de las pruebas de la proporción plasmática de aldosterona / ARP ha sido bien validada en blancos y asiáticos, no se ha validado en otros grupos raciales importantes.
INTERFERENCIA POR MEDICACIÓN
La relación de aldosterona / ARP en plasma no debe calcularse cuando el paciente toma medicamentos que pueden interferir con esta medición. La espironolactona, un antagonista del receptor de aldosterona, debe suspenderse durante 6 semanas antes de la prueba. Eplerenona, otro antagonista del receptor de aldosterona, también puede interferir con las pruebas y se debe suspender durante al menos 2 semanas antes de la prueba.
Los alfabloqueantes, como la doxazosina, no interfieren con la relación AP / ARP. Los betabloqueantes y los bloqueadores de los canales de calcio no afectan la precisión diagnóstica de la relación en la mayoría de los casos.
ARP DESPUÉS DEL AGOTAMIENTO DE LA SAL Y AGUA Y / O POSTURA ERGUIDA
En el aldosteronismo primario, la ARP es inferior a 1 ng / ml / hy no aumenta más de 2 ng / ml / h después del agotamiento de la sal y el agua, la administración de furosemida o 4 horas de postura erecta. Esta prueba, junto con las pruebas de supresión de captopril, se ha usado como una prueba de detección o como una prueba confirmatoria (de segundo nivel) para el aldosteronismo primario.
Las pruebas de confirmación se basan en el concepto de que la aldosterona se secreta de forma no regulada en el aldosteronismo primario y, por lo tanto, no puede ser suprimida por las aportaciones habituales de la regulación fisiológica. De manera similar, la ARP se suprime crónicamente y tónicamente y no se puede estimular
PRUEBAS DE SUPRESIÓN DE CAPTOPRIL Y LOSARTÁN
Esto implica la administración oral de una sola dosis de captopril (25-50 mg), un inhibidor de la ECA. En individuos sanos, los niveles de aldosterona se suprimirán a menos de 15 ng / dL. La prueba tiene una sensibilidad de 90-100% pero una especificidad de solo 50-80%.
En un estudio de 135 pacientes que se sometieron a las pruebas de captopril y losartán (bloqueador de los receptores de angiotensina-II [ARB]), Wu et al concluyeron que los valores de la relación de ARP y la concentración de aldosterona derivada de la prueba de losartan eran más precisos que los obtenidos mediante la prueba de captopril para el diagnóstico de aldosteronismo primario. [ 23 ]
Los autores encontraron que cuando se utilizaba una relación de ARP (ng / dL por ng / ml / h) superior a 35 y una concentración de aldosterona de más de 10 ng / dL, los valores de especificidad diagnóstica para captopril y losartan eran 89.1% y 93.8 %, respectivamente, y los valores de sensibilidad de diagnóstico respectivos fueron 66.2% y 84.5%. Los autores recomiendan la realización preferencial de la prueba de supresión de losartán.
PRUEBAS DE CONFIRMACIÓN (SEGUNDO NIVEL)
NIVEL DE ALDOSTERONA SÉRICA
Después de 3 días de una dieta sin restricción de sodio y 1 hora de decúbito horizontal, las personas sanas tienen niveles de aldosterona de menos de 15 ng / dL. Cuando la aldosterona sérica se eleva por encima de 22 ng / dl y se suprime la renina, la prueba de aldosterona sérica (S-Aldo) prácticamente confirma el diagnóstico de aldosteronismo primario. Sin embargo, debido a que la secreción de aldosterona es variable, el valor predictivo negativo y positivo de un único nivel aleatorio de aldosterona es limitado.
Hasta un 40% de los pacientes con aldosteronismo primario tienen niveles séricos de aldosterona que permanecen dentro del rango de referencia en las pruebas repetidas, como suele ser el caso en la hipertensión esencial (HTA). (Vea la tabla a continuación).
Algoritmo para la confirmación de aldosteronismo primario (ALGORITMO 2)
PRUEBA DE EXCRECIÓN URINARIA DE ALDOSTERONA EN 24 HORAS
La prueba de excreción urinaria de aldosterona (Aldo-U) es una de las herramientas diagnósticas confirmatorias más útiles porque es un índice para la secreción diaria total de aldosterona (de manera similar al cortisol libre de orina de 24 h [CLU]) el cual está típicamente elevado en pacientes con síndrome de Cushing).
En la mayoría de los pacientes con aldosteronismo primario, el Aldo-U de 24 horas es mayor que 14 mcg / día (después de 3 días de carga de sal). Solo alrededor del 7% de los pacientes con aldosteronismo primario tienen valores de menos de 14 mcg / día.
PRUEBA DE CARGA DE SAL
La prueba de carga de sal puede realizarse utilizando un protocolo de carga de sal intravenosa o un protocolo de carga de sal oral. El protocolo oral requiere la ingestión diaria de al menos 10-12 g de cloruro de sodio durante al menos 5 días antes de realizar la prueba. Cuando se ha cumplido el protocolo oral, se miden las excreciones Aldo-U, sodio, potasio y creatinina durante 24 horas, y se debe determinar la aldosterona sérica y la ARP. En individuos normales, el principal metabolito de Aldo-U, la aldosterona-18-glucurónido urinario, debe caer por debajo de un nivel de 17 mcg / día. La no supresión de Aldo-U-18G es altamente sugestiva de aldosteronismo primario. No obstante, esta prueba es engorrosa y rara vez se realiza.
La medición de creatinina urinaria de 24 horas valida la idoneidad de la muestra de orina, mientras que un valor de sodio urinario de 24 horas de al menos 250 mEq / día confirma una carga de sal adecuada durante los días previos a la prueba y, por lo tanto, valida las otras mediciones.
DETERMINACIÓN DE LAS PRUEBAS DEL SUBTIPO DE ALDOSTERONISMO PRIMARIO (TERCER NIVEL)
Una vez que se ha confirmado el diagnóstico de aldosteronismo primario mediante una prueba de primer o segundo nivel, el siguiente paso es determinar el subtipo de aldosteronismo primario e identificar una enfermedad curable quirúrgicamente. A efectos prácticos, esto significa distinguir entre un adenoma suprarrenal y una hiperplasia suprarrenal idiopática (HAI). (Ver el cuadro a continuación) [ 18 , 24 ]
Algoritmo para distinguir subtipos de aldosteronismo primario. (ALGORITMO 3)
En general, los pacientes con adenomas son más jóvenes que los pacientes con hiperplasia y tienen hipertensión (HTA) e hipopotasemia más severa, así como niveles más altos de aldosterona en la orina (Aldo-U) que los pacientes con HAI. Sin embargo, estos parámetros clínicos no son lo suficientemente confiables para distinguir con precisión el adenoma de la HAI
PRUEBA DE ESTIMULACIÓN POSTURAL
Los aldosteronomas se asocian con una disminución anómala del nivel de aldosterona con la postura erguida, en contraste con los pacientes con hiperplasia suprarrenal idiopática (HAI), en los que se produce un aumento del nivel de aldosterona mediado por el sistema renina angiotensina (RAS) con la postura erguida.
De forma similar, se produce un aumento del nivel de aldosterona sérica en pacientes con adenomas sensibles a renina (ASR), HTA esencial de baja renina y casos muy raros de hiperplasia suprarrenal unilateral (este último presenta características intermedias entre la hiperplasia suprarrenal idiopática [IAH] y la aldosterona). produciendo adenoma suprarrenal, ocasionalmente designado como "aldosteronismo intermedio"). [ 25 ]
Cuando la tomografía abdominal (TC) y la resonancia magnética (RM) se combinan con estimulación postural, el valor predictivo positivo (VPP) de una prueba postural anormal en la predicción de aldosteronismo primario corregible quirúrgicamente debido a un solo adenoma es del 98%.
El protocolo de prueba postural estándar implica la obtención de valores iniciales para los niveles de aldosterona sérica (Aldo-S) y actividad de renina plasmática (ARP), así como estos niveles 2 horas después de que el paciente haya asumido una postura erguida. Los niveles de Aldo-S típicamente aumentan en esta configuración al menos un 50% por encima del valor inicial en personas sanas, en personas con HTN esencial y en el subgrupo de pacientes con aldosteronismo primario que tienen hiperplasia suprarrenal idiopática (HAI) o ASR.
Entre los pacientes con aldosteronomas (adenomas productores de aldosterona, APA), los niveles de S-Aldo generalmente no aumentan o, paradójicamente, descienden a este nivel. Se ha informado que la sensibilidad y especificidad de esta prueba en el diagnóstico diferencial de las principales causas del aldosteronismo primario es tan alta como 80-85%.
PRUEBA DE ESTIMULACIÓN DE FUROSEMIDA (LASIX)
Esta prueba a menudo se combina con la prueba de postura erguida. La prueba típica implica la administración oral de 40 mg de furosemida la noche anterior, así como la mañana de la prueba. En la mañana de la prueba, después de que se haya administrado la dosis de furosemida, el paciente permanece en posición vertical de 2 a 3 horas; luego, se analizan los niveles de Aldo-S y ARP. La interpretación de los resultados de la prueba es similar a la descrita anteriormente para la prueba de estimulación postural.
RITMO DIURNO DE LA ALDOSTERONA
El ritmo circadiano de la secreción de aldosterona en individuos sanos es similar al del cortisol y depende de la corticotropina. Los valores más bajos se observan alrededor de las 11:30 p.m. hasta la medianoche, y los valores más altos ocurren temprano en la mañana alrededor de las 7: 30-8: 00 a.m. (suponiendo un ciclo normal de sueño-vigilia). Si bien esto se conserva en pacientes con aldosteronomas, generalmente se pierde en pacientes con HAI.
En algunos pacientes con aldosteronomas se pueden encontrar niveles elevados de corticosterona 18OH y / o cortisol 18OH en plasma y orina, pero son poco frecuentes en la HAI. [ 26 , 27 ]
TOMOGRAFÍA COMPUTADA Y RESONANCIA MAGNÉTICA NUCLEAR
TC
La investigación radiológica inicial en el estudio del aldosteronismo primario es una tomografía computada suprarrenal de alta resolución y corte fino (2-2.5 mm) con contraste.
Los aldosteronomas tienden a ser pequeños, en contraste con los adenomas adrenocorticales productores de cortisol; solo los aldosteronomas que tienen al menos 1 cm de diámetro se pueden detectar de manera confiable y consistente.
La sensibilidad general de la tomografía computada suprarrenal de alta resolución y corte fino es mayor al 90%, pero la imagen se complica por los muchos hallazgos falsos positivos asociados con los incidentalomas, que se informan en algunas series que se encuentran hasta en 10- 15% de la población general (su prevalencia aumenta con la edad). [ 28 ]
Además, los estudios con TC de alta resolución en realidad pueden ser perjudiciales, ya que estas exploraciones a menudo detectan la hiperplasia que acompaña a los adenomas y pueden dar lugar a una tendencia a sobrediagnosticar la hiperplasia suprarrenal idiopática (HAI). De manera similar, debido a que la hiperplasia suprarrenal a largo plazo se asocia con la formación de pseudonódulos y nódulos, esta imagen radiográfica a menudo se puede confundir con el diagnóstico de adenomas autónomos.
INDICACIONES QUIRÚRGICAS
Algunos investigadores sugieren que cuando se detecta un macroadenoma unilateral solitario (mayor de 1 cm) en una tomografía computarizada en un paciente joven en el contexto de un aldosteronismo inequívoco, está indicada la adrenalectomía unilateral. Sin embargo, debido al riesgo dependiente de la edad de que un macroadenoma suprarrenal unilateral solitario pueda ser un adenoma no funcional, algunos expertos creen que el muestreo de la vena suprarrenal [ 18 ] debe realizarse en pacientes mayores de 40 años.
RMN
En general, se acepta que la RMN no es superior a la TC con contraste para la visualización adrenal. Las tomografías computarizadas de alta resolución en realidad pueden tener una mejor definición suprarrenal.
Figura 1 Imagen de resonancia magnética (RMN) en un paciente con síndrome de Conn que muestra un adenoma suprarrenal izquierdo.
Si el examen (de TC y RMN) de un paciente con aldosteronismo primario es completamente normal, generalmente se recomienda un ensayo de tratamiento de 6 a 12 meses con antagonistas de aldosterona, después de lo cual los estudios de imágenes deben repetirse. El tratamiento con glucocorticoides también se puede considerar para el aldosteronismo remediable con glucocorticoides (GRA), si se sospecha esta condición.
GAMMAGRAFÍA NP-59 IODO-METIL-NORCOLESTEROL
Aunque es bastante difícil de configurar y no está disponible de forma rutinaria, esta prueba puede ser útil en casos seleccionados para distinguir entre adenomas e hiperplasia. En los grandes centros de referencia de medicina nuclear, el valor discriminante de la prueba se aproxima al de la muestra venosa suprarrenal (cerca del 90%), especialmente con tumores más grandes de 1,5 cm de diámetro o más. La actividad típica administrada es de 37 MBq (1 miliCurie; mCi).
Figura 2 Escintigrama obtenido mediante el uso de yodo-131-beta-yodo
El escintigrama obtenido mediante el uso de yodo-131-beta-yodometil-norcolesterol (NP-59) en un hombre de 59 años con hipertensión muestra una captación de radionúclidos bastante intensa en el tumor adrenal derecho. En la cirugía, se confirmó un tumor de Conn.
Los resultados de la prueba mejoran si ha habido una supresión previa con dexametasona de las glándulas suprarrenales usando 0,5-1 mg de dexametasona oral cada 6 horas. En este contexto, las imágenes de adenoma permanecen visibles, mientras que las imágenes de glándulas hiperplásicas se desvanecen después de 2-3 días de tratamiento con dexametasona.
La exploración estándar puede producir un resultado falso negativo para aldosteronomas pequeños; sin embargo, el rendimiento diagnóstico se puede aumentar mediante la administración conjunta de espironolactona. Esta es la principal alternativa de diagnóstico para el muestreo venoso suprarrenal (AVS).
Un estudio retrospectivo de Di Martino et al indicó que, en casos seleccionados, el aldosteronismo primario puede localizarse preoperatoriamente mediante la prueba NP-59 en lugar del muestreo venoso adrenal. Basando el rendimiento de la prueba en los resultados patológicos, la sensibilidad y el valor predictivo positivo fueron 90.9% y 83.3%, respectivamente; basar el rendimiento en el control de la presión arterial postoperatoria resultó en una sensibilidad y un valor predictivo positivo del 91,6% (para ambos). [ 29 ]
MUESTREO VENOSO SUPRARRENAL
Debido a que este procedimiento depende en gran medida de la disponibilidad de radiólogos intervencionistas técnicamente competentes, no puede (y no debe) realizarse universalmente, a pesar de que es el criterio estándar para la confirmación del exceso de aldosterona lateralizable. [ 30 , 31 , 32 ]De hecho, el muestreo de la vena suprarrenal puede realizarse selectivamente solo cuando las imágenes preoperatorias no pueden lateralizar definitivamente un aldosteronoma supuestamente unilateral. Un informe de Dekkers et al apoya esta línea de argumentación y cuestiona la noción de realizar muestreo venoso adrenal en todos los pacientes con aldosteronismo primario. El estudio encontró que en el seguimiento de un año, la intensidad de la medicación hipertensiva y los beneficios clínicos no difirieron significativamente entre los pacientes evaluados por muestreo venoso suprarrenal y los evaluados por TC suprarrenal. [ 33 ]
El muestreo venoso suprarrenal probablemente tenga su mayor utilidad cuando los hallazgos de imágenes suprarrenales son completamente normales a pesar de la evidencia bioquímica de aldosteronismo primario y en entornos en los que la patología suprarrenal bilateral está presente en las imágenes y la bioquímica sugiere la presencia de un aldosteronoma funcional.
La prueba también tiene utilidad para resolver la etiología exacta en casos de aldosteronismo primario en el que existe discordancia entre los hallazgos bioquímicos y los hallazgos radiológicos con respecto a si el aldosteronismo primario se debe a una hiperplasia suprarrenal idiopática (HAI) o un aldosteronoma.
Una serie informó que el 41% de los pacientes con una tomografía computada suprarrenal normal que tenían evidencia bioquímica de aldosteronismo primario en realidad tenían enfermedad lateralizable, mientras que el 49% de los pacientes con micronódulos bilaterales en la tomografía computarizada también tenían enfermedad lateralizable. [ 34 ] Incluso en los casos en los que solo se encontró un solo nódulo suprarrenal en las imágenes, cuando se realizó un muestreo venoso suprarrenal, confirmó una enfermedad lateralizable en solo el 51-66% de los casos.
Técnica
Las venas suprarrenales se cateterizan a través de un abordaje venoso femoral percutáneo. Los catéteres venosos derecho e izquierdo deben colocarse en las venas suprarrenales ipsilaterales para evitar errores en el manejo de las muestras. (La canulación de la vena suprarrenal derecha es técnicamente difícil debido a la corta longitud de este vaso. La vena suprarrenal izquierda es más larga, lo que permite una colocación más estable del catéter).
Al inicio y después de la estimulación con corticotropina (preferiblemente mediante infusión continua a 50 mcg / h durante la duración del estudio de muestreo), se obtienen muestras de sangre simultáneamente de ambas venas suprarrenales, así como de la vena cava inferior, y las muestras se analizan para aldosterona y cortisol.
Con el fin de documentar la colocación de los catéteres dentro de las venas suprarrenales, se calcula una relación de cortisol adrenal a vena cava (post corticotropina); debe ser mayor que 5-10.
DIAGNÓSTICO
La precisión de la prueba excede el 95% cuando el procedimiento es técnicamente exitoso. Si la secreción unilateral autónoma de aldosterona está presente en cualquier lado, la relación de las concentraciones de aldosterona entre las venas suprarrenales derecha e izquierda generalmente excede 10: 1. Se pueden presentar resultados falsos positivos cuando la estenosis de la arteria renal está presente; por lo tanto, la estenosis de la arteria renal debe excluirse completamente, especialmente antes de realizar una prueba altamente invasiva.
La mayoría de los pacientes con una fuente unilateral de aldosterona tienen relaciones de aldosterona adrenal a cortisol suprarrenales superiores a 4. Las relaciones de menos de 3 sugieren hiperplasia, y los valores de 3-4 se consideran resultados indeterminados.
Riesgos
El muestreo venoso suprarrenal no está exento de riesgos. La trombosis venosa suprarrenal e ilíaca, la hemorragia suprarrenal, la insuficiencia suprarrenal o incluso la hemorragia venosa mayor debido a desgarros transmurales y luxaciones del catéter se encuentran entre las posibles complicaciones.
ENSAYOS DE HIDROXICORTICOSTERONA Y OXOCORTISOL-HIDROXICORTISOL
Debido a las diferentes zonas adrenocorticales implicadas en la hiperplasia idiopática, en comparación con los adenomas, los ensayos de plasma 18-hidroxicorticosterona (18-OHB) o plasma y / orina 18-oxocortisol (18-oxo-F) / 18-hidroxicortisol pueden ser de uso diagnóstico [ 26 , 27 ]
5MED
Esteroides adrenocorticales de la zona de transición.
Los aldosteronomas suelen estar asociados con niveles de 18-OHB superiores a 100 ng / dL. De manera similar, el aldosteronismo remediable por glucocorticoides (GRA, hiperaldosteronismo familiar de tipo 1), aunque es una enfermedad hiperplásica por definición, también se asocia con una producción incrementada de estos derivados de 18-oxo / hidroxi. Esta distinta bioquímica no está presente en los adenomas sensibles a la renina (RRA). En este punto, pocos centros envían estas pruebas especializadas de bioquímica, y su valor actual es principalmente histórico
PRUEBA DE SUPRESIÓN DE FLUDROCORTISONA
Esta prueba funciona según el mismo principio en que se basan la infusión intravenosa de cloruro de sodio o las pruebas de carga oral de sal para confirmar el diagnóstico de aldosteronismo primario. Sin embargo, la prueba de supresión de la fludrocortisona ha perdido gran parte de su popularidad, ya que requiere hospitalización del paciente y de 4 a 5 días para completarse, y actualmente es principalmente de interés histórico.
Por razones de compleción, la descripción de esta prueba es la siguiente: la fludrocortisona se administra por vía oral a una dosis de 0.1-0.2 mg cada 6 horas, junto con cloruro de sodio y potasio suplementarios. En el individuo sano, después de esta estimulación, el nivel sérico de aldosterona (S-Aldo) se suprime típicamente a menos de 8 ng / dl, con una excreción urinaria de aldosterona (U-Aldo) menor de 12 mcg / día.
En pacientes con aldosteronismo primario, ni el nivel de aldosterona en orina ni el nivel de aldosterona en plasma suprime los umbrales mencionados anteriormente.
PRUEBA DE SUPRESIÓN DE DEXAMETASONA
Esta prueba es relevante solo en el contexto de un posible aldosteronismo familiar. Habitualmente, en pacientes con aldosteronismo primario, la dexametasona se asocia con una reducción transitoria de leve a moderada de los niveles de aldosterona en plasma y urinaria, aunque no en el rango de referencia normal. [ 7 ]
En el subgrupo de pacientes con aldosteronismo primario con aldosteronismo remediable con glucocorticoides (GRA), dosis pequeñas de dexametasona (1-2 mg / día) inducen la normalización completa en los niveles plasmáticos y de aldosterona en la orina. Esto está invariablemente asociado con una mejoría en la hipertensión (HTA) en estos pacientes. Otros informes sugieren un nivel de corte para la aldosterona plasmática de menos de 4 ng / dL y / o una supresión relativa de aldosterona en plasma de más del 80% de la línea de base para el diagnóstico de GRA después del desafío con dexametasona.
Existen tres variantes del aldosteronismo primario familiar. El aldosteronismo primario familiar tipo 1 (también llamado GRA) se relaciona con la mejoría de la HTA con dosis bajas de dexametasona. Los aldosteronismos primarios familiares tipo 2 y 3 no son suprimibles con dexametasona.
PRUEBA DE METOCLOPRAMIDA (REGLAN)
Esta es una prueba no invasiva para distinguir entre aldosteronomas e hiperplasia suprarrenal idiopática (IAH). Aprovecha la expresión diferencial de los receptores de dopamina en la membrana celular de las células adrenocorticales.
En condiciones normales, la dopamina causa la inhibición tónica de la aldosterona, mientras que la serotonina (5-HT) provoca una mayor secreción de aldosterona in vivo. La metoclopramida es un antagonista del receptor de la dopamina D2, así como un agonista parcial del receptor 4 de la serotonina (5-HT4) y, por lo tanto, su administración conduce a un aumento de los niveles de aldosterona. Este patrón normal de respuesta se retiene en pacientes con aldosteronomas e hipertensión de baja renina (HTN), pero no en pacientes con HAI. [ 35 ]
Después de una inyección intravenosa de 10 mg de metoclopramida, los niveles séricos de aldosterona aumentan significativamente en pacientes con aldosteronomas, pero permanecen sin cambios o paradójicamente reducidos en pacientes con HIA. Esta prueba experimentó un aumento en el uso a mediados de la década de 1990, especialmente en Europa, pero actualmente se usa muy poco en los Estados Unidos.
ESTUDIOS DE LABORATORIO ADICIONALES
PRUEBA DE ESTIMULACIÓN DE CORTICOTROPINA
La prueba de estimulación con corticotropina usa la inyección intravenosa estándar de 250 mcg. En aldosteronomas, típicamente se observa una respuesta robusta de aldosterona. En la hiperplasia suprarrenal idiopática (HAI), la oleada de aldosterona es considerablemente más débil. La prueba ya no se usa porque el valor discriminante de la prueba es bastante pobre. [ 6
PRUEBA DE INFUSIÓN DE ANGIOTENSINA II
Esta prueba implica evaluar la respuesta de la actividad de la renina plasmática (ARP) y la aldosterona sérica (Aldo-S) a una infusión continua de angiotensina II. Las características de respuesta son similares a las observadas en las pruebas de postura, con un aumento apropiado en el nivel de aldosterona observado en HAI pero no en aldosteronomas.
La prueba de infusión de angiotensina II es mucho menos popular que otras pruebas de aldosteronismo porque requiere infusión continua y monitorización hemodinámica cercana, y rara vez se usa hoy en día.
HALLAZGOS HISTOLÓGICOS
Los hallazgos histológicos varían de acuerdo con el tipo de aldosteronismo primario que tiene un paciente. Los aldosteronomas típicos se caracterizan por tejido adenomatoso, generalmente con morfología de tipo zona fasciculada. La mayoría de estos tumores son pequeños (<3 a="" acinos="" asociada="" c="" cargadas="" cm="" cordones.="" de="" di="" difusa="" dispuestas="" en="" focal="" hiperplasia="" l="" la="" las="" lulas="" mayor="" menudo="" metro="" o:p="" o="" ocurre.="" pidos="" son="" y="">3>
Los adenomas sensibles a la renina (RRA) se caracterizan por una morfología de tipo zona glomerulosa. Las únicas otras características distintivas son características bioquímicas únicas y predecibles.
La hiperplasia suprarrenal idiopática (IAH) se caracteriza por hiperplasia difusa que puede ser micronodular, macronodular o una mezcla de ambas. La morfología de las células es comúnmente similar a la zona glomerulosa.
En la hiperplasia suprarrenal primaria (HAP), típicamente se observa hiperplasia difusa, que es unilateral y tiene morfología de la zona fasciculada.
El síndrome de aldosteronismo primario rara vez ocurre en el contexto del carcinoma suprarrenal, por lo que la causa de la hipersecreción de aldosterona es una enfermedad maligna (en lugar de un adenoma benigno). En tales casos, la histopatología revela las características típicas del carcinoma adrenocortical (CCA) , incluidas las figuras mitóticas, la invasión local y las metástasis ganglionares locorregionales.
TRATAMIENTO
Consideraciones de enfoque
Entre los principales objetivos del tratamiento para el aldosteronismo primario se encuentran (1) la normalización de la presión arterial, (2) la normalización de los niveles de potasio sérico y otros electrolitos, y (3) la normalización de los niveles séricos de aldosterona. [ 18 , 36 ]
El tratamiento apropiado para el aldosteronismo primario depende de su causa. Aunque la hipertensión (HTA) se cura frecuentemente después de una adrenalectomía unilateral en pacientes con aldosteronismo primario secundario a un aldosteronoma suprarrenal, la tasa media de curación de la HTA es solo del 19% después de una suprarrenalectomía unilateral o bilateral en pacientes con hiperplasia suprarrenal idiopática (HAI). Por lo tanto, el tratamiento médico es el tratamiento de elección para la variante HAI de aldosteronismo primario.
MED
Efectos de los principales antihipertensivos en el sistema renina-angiotensina.
En pacientes con un aldosteronoma, el tratamiento médico se usa preoperatoriamente para controlar la presión arterial y corregir la hipocalemia, disminuyendo así el riesgo quirúrgico. La terapia médica también se administra a pacientes con HTA persistente en el postoperatorio, candidatos quirúrgicos pobres y pacientes que rechazan la cirugía.
En pacientes con insuficiencia cardíaca que tienen aldosteronismo secundario, se ha demostrado que el antagonismo de la aldosterona por espironolactona y eplerenona confiere un beneficio de supervivencia. [ 37 ]
TERAPIA FARMACOLÓGICA
BLOQUEADORES DE LOS CANALES DE CALCIO
Al inhibir el flujo de calcio intracelular en las células adrenocorticales, los bloqueadores de los canales de calcio dihidropiridínicos reducen la producción de aldosterona en respuesta a una variedad de estimulantes, que incluyen potasio, corticotropina y angiotensina-II.
La nifedipina es la más ampliamente estudiada de estos medicamentos; sin embargo, aunque la nifedipina causa una mejoría significativa en pacientes con hipertensión (HTA), no aborda la fisiopatología de la afección. La actividad de la renina plasmática (ARP), los niveles de aldosterona, el volumen plasmático y las concentraciones séricas de potasio permanecen esencialmente sin cambios mientras se usa nifedipina.
ANTAGONISTAS DE MINERALOCORTICOIDES
Los antagonistas de mineralocorticoides, como la espironolactona, son ligandos inhibidores del receptor mineralocorticoide (MR). Logran un notable control de la presión arterial y la normalización del volumen plasmático y las concentraciones séricas de potasio, particularmente en pacientes con aldosteronomas. [ 38 ]
Los efectos saludables de la espironolactona parecen deberse principalmente a su impacto en el equilibrio de la sal y el agua en lugar de su antagonismo de la aldosterona en el riñón. La combinación de espironolactona y tiazidas a menudo proporciona un mejor control de la presión arterial que la espironolactona sola.
Debido a los efectos adversos estrógenos de la espironolactona, incluida la impotencia y la ginecomastia, el incentivo para desarrollar un agente antialdosterónico de eficacia similar sin estos efectos adversos es considerable. La eplerenona es un agente antialdosterónico selectivo que puede cumplir esta promesa, ya que es un antagonista específico del receptor de la aldosterona que no tiene los efectos antiandrógenos adicionales asociados con la espironolactona.
GLUCOCORTICOIDES
En el subgrupo de pacientes con aldosteronismo remediable con glucocorticoides (GRA), el tratamiento de elección es la administración de la dosis más baja posible de glucocorticoides que se puede usar para lograr un control adecuado de la presión arterial. Debido a los posibles efectos adversos que pueden resultar incluso de un exceso sutil de glucocorticoides, el uso de glucocorticoides de acción corta, como la prednisona y la hidrocortisona (en lugar de la dexametasona), generalmente es lo mejor.
OTROS
Los inhibidores de la ECA y los bloqueadores de los receptores de angiotensina (BRA) también son opciones de tratamiento potenciales. Las opciones de tratamiento médico menos ideales incluyen diuréticos ahorradores de potasio, como triamtereno y amilorida, que no son antagonistas de mineralocorticoides. La amilorida actúa a nivel del túbulo contorneado distal pero no se une a los receptores mineralocorticoides.
Existen algunos informes del uso de inyección percutánea de etanol o ácido acético en aldosteronomas como una modalidad de tratamiento; en estos casos, el tratamiento generalmente se administró a pacientes para quienes la cirugía estaba contraindicada. [ 39 ] Esta técnica no es popular ni está bien validada. Además, requiere la experiencia técnica de un radiólogo intervencionista altamente capacitado.
CONSIDERACIONES
La terapia no quirúrgica también es una opción de tratamiento viable en pacientes que tienen enfermedad lateralizable pero que son candidatos quirúrgicos pobres debido a otras comorbilidades coexistentes. También es una opción de tratamiento viable en el raro entorno de los adenomas suprarrenales funcionales bilaterales que de otra manera requerirían una adrenalectomía bilateral.
CIRUGÍA SUPRARRENAL
La cirugía es el tratamiento de elección para las variantes lateralizables del aldosteronismo primario, incluidos los aldosteronomas típicos, los adenomas sensibles a la renina (RRA) y la hiperplasia suprarrenal primaria (HAP).
Preoperatoriamente, una vez que se hayan realizado y confirmado los diagnósticos bioquímicos y anatómicos, el paciente debe comenzar un ciclo de espironolactona de 3 a 5 semanas. Esto sirve como una herramienta de diagnóstico adicional (para confirmar el diagnóstico de aldosteronismo primario) y como un medio para predecir la respuesta de la presión arterial que se puede esperar después de la cirugía.
Una adrenalectomía se puede realizar a través de una laparotomía formal o mediante el uso de una técnica laparoscópica (con el rendimiento de este último cada vez más común). La opción laparoscópica ahora permite ofrecer terapia quirúrgica a pacientes relativamente débiles que no podrían resistir una laparotomía formal. Los estudios en curso evalúan sistemáticamente el lugar de las operaciones de conservación suprarrenal frente a la adrenalectomía unilateral total en estos pacientes. [ 40 ]
Entre las opciones estudiadas se encuentran (1) suprarrenalectomía parcial, en la que se realiza una resección en cuña de la glándula con el adenoma junto con enucleación de aldosteronoma y (2) adrenalectomía ahorradora de médula, en la que se intenta retener la médula suprarrenal tejido mientras se elimina la corteza.
RESULTADOS
Alrededor del 60-70% de los pacientes se vuelven normotensos después de la cirugía curativa para aldosteronomas cuando se evalúan 1 año después de la operación. La hipertensión (HTA) típicamente no se resuelve inmediatamente después de la operación sino, más bien, durante 3 a 6 meses. (Aunque después de la cirugía, prácticamente todos los pacientes con un aldosteronoma tienen reducciones significativas en la secreción de aldosterona y la presión arterial y también demuestran una corrección de la hipopotasemia. [ 41 , 42 , 43 , 44 , 45 ] )
Un estudio retrospectivo de 168 pacientes con aldosteronismo primario sometidos a una suprarrenalectomía encontró que la HTA se curó o controló en el 77% de los pacientes con adenoma unilateral y en el 68% de los pacientes con aldosteronismo que no tenían adenoma, pero cuya relación aldosterona / cortisol fue al menos 5 veces mayor en el lado dominante que en el lado no dominante. [ 43 ]
El porcentaje de pacientes que permanecen normotensos 5 años después de la operación es aproximadamente del 53%. La resolución de la HTN después de la adrenalectomía ocurre invariablemente en el contexto de un historial familiar en el que la HTN está ausente y / o cuando el paciente ha usado preoperatoriamente 2 o menos antihipertensivos.
La adrenalectomía tiene muy poca utilidad en el contexto de la hiperplasia suprarrenal idiopática (HAI). En los casos informados en los que se ha realizado cirugía involuntariamente en pacientes con IAH, los efectos sobre la presión arterial, la hipocalemia y la hipersecreción de aldosterona han sido mínimos, lo que refuerza aún más la necesidad de hacer un diagnóstico correcto antes de justificar una adrenalectomía.
La persistencia de la HTA después del aparente tratamiento quirúrgico de la enfermedad lateralizable es más común en pacientes mayores de 45 años, en aquellos que tuvieron HTA durante más de 5 años antes de la cirugía y en personas que no respondieron antes de la operación a la espironolactona. [ 43 , 44 ]
La HTA persistente puede estar relacionada con el restablecimiento de los barorreceptores, los cambios hemodinámicos establecidos, los cambios estructurales en los vasos sanguíneos o la HTA esencial coincidente.
Otra posibilidad a considerar en la HTA persistente es una resección incompleta del adenoma, con tejido remanente hiperplásico remanente. La coexistencia de nefroesclerosis hipertensiva en algunos pacientes con HTN persistente también es una posibilidad clara. También se debe considerar la coexistencia de otras causas secundarias de HTA; la estenosis de la arteria renal es una consideración importante.
CUIDADO PREOPERATORIO Y POSTOPERATORIO
Antes de la cirugía, los pacientes deben recibir al menos 8-10 semanas de tratamiento médico para disminuir la presión arterial y corregir los síndromes metabólicos que a menudo se asocian con el aldosteronismo primario.
En el postoperatorio, los perfiles metabólicos deben ser monitoreados de cerca. La mayoría de los pacientes no desarrollan hipomineralocorticoidismo permanente y, por lo tanto, no requieren reemplazo de fludrocortisona.
Para los pacientes que desarrollan hipoaldosteronismo, los síntomas pueden persistir por un tiempo prolongado y pueden ser similares al retraso observado en la recuperación de glucocorticoides suprarrenales luego de la supresión crónica de la corticotropina por esteroides exógenos. Sin embargo, si se desarrolla hiperpotasemia significativa , se deben suspender los suplementos de potasio y se puede iniciar el tratamiento con furosemida a dosis de 80 a 160 mg diarios.
DIETA
Una dieta baja en sal, aunque útil para lograr el control de la presión arterial en el aldosteronismo primario, puede estar asociada con resultados negativos falsos en las pruebas bioquímicas.
Una dieta alta en sal dificulta el logro del control de la presión arterial y puede dar resultados falsos positivos en las pruebas bioquímicas.
RESUMEN DE PAUTAS
Una serie de recomendaciones sobre el manejo del aldosteronismo primario fueron desarrolladas por la Sociedad Francesa de Endocrinología (SFE), la Sociedad Francesa de Hipertensión (SFHTA) y la Asociación de Cirugía Endocrina Francófona (AFCE). [ 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 ] Incluyen una recomendación para detectar aldosteronismo primario en pacientes con cualquiera de los siguientes:
Hipertensión severa (presión arterial sistólica de 180 mm Hg o mayor o presión arterial diastólica de 110 mm Hg o mayor)
Hipertensión resistente (presión arterial sistólica de 140 mm Hg o superior o presión arterial diastólica de 90 mm Hg o superior, incluso después del uso de al menos tres agentes antihipertensivos, incluido un diurético tiazídico)
Hipertensión asociada a hipopotasemia (ya sea espontánea o diurética asociada)
Hipertensión o hipocalemia relacionada con un incidentaloma suprarrenal
Con respecto a la cirugía, las recomendaciones establecen que la adrenalectomía laparoscópica se debe utilizar en pacientes con aldosteronismo primario lateralizado que son candidatos para cirugía. En términos de tratamiento médico, se recomienda el uso de amilorida en pacientes con intolerancia a la espironolactona, mientras que la eplerenona se sugiere como otra alternativa en casos de intolerancia a la espironolactona o si la amilorida no controla suficientemente la hipertensión.
En una actualización de 2016 de sus guías de práctica clínica para el diagnóstico y tratamiento del aldosteronismo primario de 2008, la Endocrine Society incluyó las siguientes recomendaciones [ 54 , 55 ] :
Detección de la condición en sujetos con presión arterial sostenida superior a 150/100 mm Hg, como se encuentra en cada una de las tres mediciones obtenidas en días diferentes, así como en pacientes con hipertensión (presión arterial> 140/90 mm Hg) resistente a tres convencionales medicamentos antihipertensivos (incluido un diurético) o con presión arterial controlada (<140 a="" accidente="" aldosteronismo="" antecedentes="" antihipertensivos="" apnea="" cerebrovascular="" con="" cuatro="" de="" debe="" del="" diur="" e="" edad="" el="" en="" espont="" familiares="" grado="" hg="" hipertensi="" hipertensos="" hipocalemia="" incidentaloma="" inducida="" inicio="" los="" m="" medicamentos="" mm="" n="" neos="" o:p="" o="" os="" pacientes="" por="" primario="" primer="" realizar="" s="" se="" sue="" suprarrenal="" tambi="" tamizaje="" temprana="" temprano="" ticos="" todos="" una="" y="">140>
El uso de la relación aldosterona / renina plasmáticas para la detección de aldosteronismo primario posible en los grupos de pacientes anteriores
El uso de una o más pruebas confirmatorias en pacientes con una relación plasmática positiva de aldosterona / renina para confirmar o excluir definitivamente el diagnóstico
El uso de tomografía computarizada adrenal en todos los pacientes con aldosteronismo primario para excluir grandes masas que pueden representar el carcinoma adrenocortical y para ayudar al radiólogo intervencionista y al cirujano donde sea apropiado anatómicamente
Cuando el tratamiento quirúrgico es factible y deseado por el paciente, el uso del muestreo venoso suprarrenal para hacer la distinción entre enfermedad suprarrenal unilateral y bilateral
El uso de pruebas genéticas para el hiperaldosteronismo familiar tipo 1 en pacientes en los que se confirma el inicio del aldosteronismo primario antes de los 20 años y en aquellos con antecedentes familiares de aldosteronismo primario o accidente cerebrovascular a una edad temprana (<40 a="" o:p="" os="">40>
El uso de adrenalectomía laparoscópica unilateral en pacientes con aldosteronismo primario unilateral documentado (es decir, adenoma productor de aldosterona o hiperplasia suprarrenal unilateral) o, en pacientes que no pueden o no desean someterse a cirugía, la administración de tratamiento médico que incluye un antagonista del receptor mineralocorticoide
La administración de tratamiento médico con un antagonista del receptor de mineralocorticoides en pacientes con aldosteronismo primario debido a enfermedad suprarrenal bilateral
En pacientes con aldosteronismo remediable con glucocorticoides, la administración de la dosis más baja de glucocorticoides para reducir la hormona adrenocorticotrópica y así normalizar la presión arterial y los niveles de potasio como tratamiento de primera línea; si la presión sanguínea no se normaliza con el glucocorticoide solo, se puede agregar un antagonista del receptor mineralocorticoide
RESUMEN DE MEDICACIÓN
En el aldosteronismo primario no quirúrgico, el tratamiento médico es el tratamiento de elección. El fármaco que es el tratamiento de primera elección para la mayoría de las variantes del aldosteronismo primario no quirúrgico es la espironolactona, que se usa para lograr el normoaldosteronismo y para ayudar con el control de la presión arterial. Los suplementos de potasio no deben administrarse de forma rutinaria con espironolactona debido a la posibilidad de desarrollar hipercalemia.
En pacientes que no pueden tolerar la espironolactona, se pueden utilizar otros diuréticos ahorradores de potasio, como la amilorida y el triamtereno, aunque se consideran opciones menos ideales. [ 37 ]
El aldosteronismo remediable con glucocorticoides (GRA) se trata con pequeñas dosis de glucocorticosteroides (es decir, hidrocortisona, prednisona). En dosis óptimas, los glucocorticosteroides normalizan la aldosterona y la presión arterial.
Se pueden agregar varios antihipertensivos para lograr un control adecuado de la presión arterial. Los bloqueadores de los canales de calcio dihidropiridínicos (p. Ej., Nifedipina) inhiben directamente la producción de aldosterona; sin embargo, aunque producen mejorías significativas en pacientes con hipertensión (HTA), no abordan la fisiopatología. La actividad de la renina plasmática (PRA), los niveles de aldosterona, el volumen plasmático y las concentraciones séricas de potasio permanecen esencialmente sin cambios con el uso de nifedipina.
Otros agentes de segundo paso para el control de la presión arterial incluyen los diuréticos tiazídicos, los inhibidores de la enzima convertidora de la angiotensina (ECA) y los bloqueadores del receptor de la angiotensina II. [ 23 ]
Una dieta restringida en sodio (menos de 80 mEq o menos de2 g de sodio por día), el mantenimiento del peso corporal ideal y el ejercicio aeróbico regular contribuyen sustancialmente al éxito del tratamiento farmacológico.
ANTAGONISTAS DE ALDOSTERONA, SELECTIVOS
Estos agentes compiten con los sitios receptores de aldosterona, reduciendo el edema y la ascitis.
ESPIRONOLACTONA (ALDACTONE)
La espironolactona se une competitivamente a los receptores en el sitio de intercambio sodio-potasio dependiente de aldosterona en el túbulo renal contorneado distal. Proporciona efectos diuréticos y antihipertensivos, lo que provoca una mayor excreción de sodio y agua, al tiempo que conserva el potasio. La espironolactona se administra sola o con un agente diurético que actúa sobre el túbulo renal proximal. La espironolactona puede bloquear los efectos de la aldosterona en los músculos lisos arteriolares.
EPLERENONA (INSPRA)
La eplerenona bloquea selectivamente la aldosterona en los receptores mineralocorticoides en los tejidos epiteliales (p. Ej., Riñones) y no epiteliales (p. Ej., Corazón, vasos sanguíneos, cerebro), disminuyendo así la presión arterial y la reabsorción de sodio.
DIURÉTICOS AHORRADORES DE POTASIO
Estos agentes se usan como medicamentos de segunda línea para el tratamiento del aldosteronismo primario debido a una enfermedad no lateralizante y / o enfermedad de lateralización para la cual la cirugía está contraindicada o rechazada. A menudo se deben usar con otros antihipertensivos para lograr el mejor control de la presión arterial, ya que no son antihipertensivos potentes.
TRIAMTERENE (DYRENIUM)
El triamtereno es un diurético ahorrador de potasio con propiedades natriuréticas relativamente débiles. Ejerce un efecto diurético en el túbulo renal distal para inhibir la reabsorción de sodio a cambio de potasio e hidrógeno. El triamtereno aumenta la excreción de sodio y reduce la pérdida excesiva de potasio e hidrógeno asociada con hidroclorotiazida. No es un antagonista competitivo de mineralocorticoides; su efecto conservador de potasio se observa en pacientes con enfermedad de Addison (es decir, sin aldosterona).
El inicio y la duración de la actividad con triamtereno son similares a los de hidroclorotiazida. No se demuestra un efecto antihipertensivo predecible. Se absorbe rápidamente después de la administración oral, y los niveles plasmáticos máximos se alcanzan dentro de 1 hora de la dosificación. El triamtereno se metaboliza principalmente a un conjugado de sulfato de hidroxitriamtereno. Los niveles plasmáticos y urinarios de este metabolito superan con creces los niveles de triamtereno.
AMILORIDA
Amilorida es una pirazina-carbonil-guanidina no relacionada químicamente con otros agentes antiqualiuréticos o diuréticos conocidos. Es un fármaco conservador del potasio (antiacaliurético) que, en comparación con los diuréticos tiazídicos, posee actividad natriurética, diurética y antihipertensiva débil. Los efectos de Amiloride han sido parcialmente aditivos a los efectos de los diuréticos tiazídicos en algunos estudios clínicos. Cuando se administra con una tiazida o diurético de asa, se ha demostrado que disminuye la excreción urinaria aumentada de magnesio que ocurre cuando se usa una tiazida o un diurético de asa solo.
La amilorida tiene actividad conservadora de potasio en pacientes que reciben agentes diuréticos kaliuréticos. Amiloride no es un antagonista de la aldosterona, y sus efectos se observan en ausencia de aldosterona. Ejerce su efecto ahorrador de potasio a través de la inhibición de la reabsorción de sodio en el túbulo contorneado distal, el túbulo colector cortical y el conducto colector. Esto disminuye el potencial negativo neto de la luz tubular y reduce la secreción de potasio e hidrógeno y sus excreciones posteriores.
Amiloride por lo general comienza a actuar dentro de las 2 horas después de una dosis oral. Su efecto sobre la excreción de electrolitos alcanza un pico entre 6-10 horas y dura aproximadamente 24 horas. Los niveles plasmáticos máximos se obtienen en 3-4 horas, y la vida media plasmática del fármaco varía entre 6 y 9 horas.
Amiloride no es metabolizado por el hígado; en cambio es excretado sin cambios por los riñones. Dentro de las 72 horas, aproximadamente el 50% de una dosis de amilorida se excreta en la orina y el 40% en las heces. El medicamento tiene poco efecto sobre la tasa de filtración glomerular o sobre el flujo sanguíneo renal. Debido a que el hígado no metaboliza el hidrocloruro de amilorida, no se prevé la acumulación de medicamentos en pacientes con disfunción hepática; sin embargo, la acumulación puede ocurrir si se desarrolla el síndrome hepatorrenal.
Amiloride rara vez debe usarse solo. Usados como agentes únicos, los diuréticos ahorradores de potasio, como la amilorida, aumentan el riesgo de hipercalemia (aproximadamente 10% con amilorida). Amiloride debe usarse solo solo cuando se haya documentado la hipocalemia persistente y solo con una titulación cuidadosa de la dosis y una estrecha monitorización de los niveles séricos de electrolitos.
DIURÉTICOS TIACÍDICOS
Los diuréticos tiazídicos inhiben la reabsorción de sodio en los túbulos distales, aumentando la excreción de sodio, agua y iones de potasio e hidrógeno. Han sido efectivos en el tratamiento de la hipertensión de diversas etiologías. Además de disminuir la reabsorción de sodio, también parecen disminuir la sensibilidad de los vasos sanguíneos a las sustancias vasopresoras circulantes. En todos los pacientes tratados con diuréticos, se deben controlar los niveles de electrolitos. Los ejemplos de diuréticos tiazídicos son hidroclorotiazida y clortalidona.
HIDROCLOROTIAZIDA (MICROZIDE)
La hidroclorotiazida inhibe la reabsorción de sodio en los túbulos distales, causando una mayor excreción de sodio y agua, así como de iones de potasio e hidrógeno.
CLORTALIDONA
La clortalidona inhibe la reabsorción de sodio en los túbulos distales, causando una mayor excreción de sodio y agua, así como de iones de potasio e hidrógeno.
BLOQUEADORES DE LOS CANALES DE CALCIO
Los bloqueadores de los canales de calcio afectan la presión arterial al disminuir la resistencia periférica vascular. Con los bloqueadores de los canales de calcio de acción corta, la respuesta cardíaca a esta acción es variable y puede producirse taquicardia. Las preparaciones de acción prolongada pueden causar una disminución en la frecuencia cardíaca.
Los bloqueadores de los canales de calcio se clasifican por su estructura y tienen diferentes grados de selectividad en sus efectos sobre el músculo liso vascular. Las dihidropiridinas no ejercen efectos electrofisiológicos y se usan comúnmente para controlar la hipertensión. Se puede producir rubor facial. Los ejemplos de bloqueadores de los canales de calcio incluyen amlodipina e isradipina.
AMLODIPINA (AMLOC)
La amlodipina se considera generalmente como una dihidropiridina, aunque la evidencia experimental sugiere que también se puede unir a sitios de unión a no dihidropiridina. Es apropiado para la profilaxis de la angina variante y tiene efectos antianginosos y antihipertensivos. La amlodipina bloquea la liberación postexcita de iones de calcio en el músculo liso vascular y cardíaco, inhibiendo así la acción de la adenosina trifosfatasa (ATPasa) en la contracción de miofibrillas.
El efecto global de la amlodipina es la reducción de los niveles de calcio intracelular en las células cardíacas y de músculo liso de la vasculatura coronaria y periférica, lo que da como resultado la dilatación de las arterias coronarias y periféricas. La amlodipina también aumenta el suministro de oxígeno al miocardio en pacientes con angina vasoespástica y puede potenciar los efectos del inhibidor de la enzima convertidora de la angiotensina (ECA).
Durante la despolarización, la amlodipina inhibe la entrada de iones de calcio en canales lentos y áreas sensibles al voltaje del músculo liso vascular y el miocardio. Beneficia a pacientes no embarazadas con disfunción sistólica, hipertensión o arritmias. Tiene una vida media sustancialmente más larga que nifedipina y diltiazem y se administra una vez al día.
FELODIPINO
La felodipina relaja el músculo liso coronario y produce vasodilatación coronaria, que a su vez mejora el suministro de oxígeno al miocardio. Beneficia a pacientes no embarazadas con disfunción sistólica, hipertensión o arritmias. Puede usarse durante el embarazo si está clínicamente indicado.
Los bloqueadores de los canales de calcio potencian los efectos del inhibidor de la ECA. La protección renal no está probada, pero estos agentes reducen las tasas de morbilidad y mortalidad en la insuficiencia cardíaca congestiva. Los bloqueadores de los canales de calcio están indicados en pacientes con disfunción diastólica. Son efectivos como monoterapia en pacientes negros y ancianos.
ISRADIPINA
Isradipina es un bloqueador de canales de calcio dihidropiridina. Inhibe la entrada de calcio en áreas sensibles al voltaje seleccionadas del músculo liso vascular y el miocardio durante la despolarización. Esto causa la relajación del músculo liso vascular coronario, lo que produce una vasodilatación coronaria. La vasodilatación reduce la resistencia sistémica y la presión arterial, con un pequeño aumento en la frecuencia cardíaca en reposo. Isradipina también tiene efectos inotrópicos negativos.
NIFEDIPINA, DE LIBERACIÓN PROLONGADA (ADALAT CC, NIFEDICAL XL, PROCARDIA XL)
La nifedipina de liberación prolongada relaja el músculo liso coronario y produce vasodilatación coronaria, que a su vez mejora el suministro de oxígeno al miocardio.
INHIBIDORES DE ECA
Los inhibidores de la enzima convertidora de la angiotensina (ECA) previenen la conversión de angiotensina I a angiotensina II, un potente vasoconstrictor y una menor secreción de aldosterona. Son fármacos efectivos y bien tolerados sin efectos adversos sobre los niveles de lípidos en plasma o la tolerancia a la glucosa. Impiden la progresión de la nefropatía diabética y otras formas de glomerulopatías, pero parecen ser menos eficaces en pacientes negros que en pacientes blancos. Los inhibidores de la ECA están contraindicados en el embarazo.
Los pacientes con alta actividad de renina plasmática (ARP) pueden tener una respuesta hipotensora excesiva a los inhibidores de la ECA. Los pacientes con vasculopatía renal bilateral o con un solo riñón, cuya perfusión renal se mantiene con altos niveles de angiotensina II, pueden desarrollar insuficiencia renal aguda irreversible cuando se los trata con inhibidores de la ECA, y se debe tener precaución con su uso en estos pacientes. Curiosamente, aunque el aldosteronismo primario es una afección asociada con baja renina plasmática, la secreción de aldosterona parece ser exquisitamente sensible incluso a concentraciones subnormales de angiotensina II. Este fenómeno parece ser la base de la eficacia de los inhibidores de la ECA en el aldosteronismo primario (específicamente hiperplasia suprarrenal idiopática [AHI]).
La tos y el angioedema son menos comunes con los miembros más nuevos de esta clase que con el captopril. Deben controlarse las concentraciones séricas de potasio y creatinina sérica para el desarrollo de hipercalemia y azotemia. Los agentes en esta clase incluyen captopril, lisinopril y enalapril.
CAPTOPRIL
El captopril previene la conversión de angiotensina I a angiotensina II, un potente vasoconstrictor, lo que resulta en una menor secreción de aldosterona. Se absorbe rápidamente, pero la biodisponibilidad se reduce significativamente con la ingesta de alimentos. Captopril logra una concentración máxima en 1 hora y tiene una vida media corta. La droga es eliminada por el riñón; la función renal deteriorada requiere la reducción de la dosis. Captopril se absorbe bien por vía oral.
Administre captopril al menos 1 hora antes de las comidas. Si se agrega al agua, úselo en 15 minutos. La dosis puede ser baja al principio, luego ajustarse según sea necesario y según lo tolere el paciente.
ENALAPRIL (VASOTEC)
Enalapril previene la conversión de angiotensina I a angiotensina II, un potente vasoconstrictor, lo que resulta en niveles elevados de renina plasmática y una reducción en la secreción de aldosterona. El medicamento ayuda a controlar la presión sanguínea y la proteinuria. El enalapril disminuye la relación de flujo pulmonar a sistémico en el laboratorio de cateterización y aumenta el flujo sanguíneo sistémico en pacientes con resistencia vascular pulmonar relativamente baja.
Enalapril tiene un efecto clínico favorable cuando se administra durante un período prolongado. Debido a que ayuda a prevenir la pérdida de potasio en los túbulos distales, el enalapril reduce la cantidad de suplementos de potasio oral que necesita el paciente.
LISINOPRIL (PRINIVIL, ZESTRIL)
Lisinopril previene la conversión de angiotensina I a angiotensina II, un vasoconstrictor potente, lo que resulta en niveles elevados de renina en plasma y una reducción en la secreción de aldosterona.
BENAZEPRIL
Benazepril previene la conversión de angiotensina I a angiotensina II, un vasoconstrictor potente, lo que resulta en niveles elevados de renina plasmática y una reducción en la secreción de aldosterona.
Cuando los pacientes pediátricos no pueden tragar las tabletas o la dosis calculada no se corresponde con la concentración de la tableta, se puede combinar una suspensión extemporánea. Combine 300 mg (15 tabletas de 20 mg de concentración) en 75 ml de vehículo de suspensión Ora-Plus y agite bien durante al menos 2 minutos. Deje que las tabletas se sientan y se disuelvan durante al menos 1 hora, luego agite de nuevo durante 1 minuto. Agregue 75 ml de Ora-Sweet. La concentración final es de 2 mg / ml, con un volumen total de 150 ml. El tiempo de caducidad es de 30 días con refrigeración.
FOSINOPRIL
Fosinopril es un inhibidor de la ECA competitivo. Previene la conversión de angiotensina I a angiotensina II, un potente vasoconstrictor, lo que resulta en niveles elevados de renina en plasma y una reducción en la secreción de aldosterona. Disminuye la presión intraglomerular y la filtración de proteína glomerular al disminuir la constricción arteriolar eferente.
QUINAPRIL
Quinapril es un inhibidor de la ECA competitivo. Reduce los niveles de angiotensina II, disminuyendo la secreción de aldosterona.
RAMIPRIL (TRITACE)
Ramipril inhibe inhibe parcialmente tanto la actividad de la ECA circulante como del tejido, por lo tanto, reduce la formación de angiotensina II en el tejido y el plasma. Ramipril tiene un efecto antihipertensivo incluso en pacientes con hipertensión de baja renina.
BLOQUEADORES DEL RECEPTOR DE ANGIOTENSINA II
Los bloqueadores de los receptores de la angiotensina II reducen la presión sanguínea al bloquear el receptor final (es decir, la angiotensina II) en el eje renina-angiotensina. Al igual que los inhibidores de la enzima convertidora de la angiotensina (ECA), están contraindicados en el embarazo. Deben controlarse los niveles séricos de electrolitos y creatinina. Irbesartan y losartán son ejemplos de bloqueadores del receptor de angiotensina II (BRA). Curiosamente, aunque el aldosteronismo primario es una afección asociada con baja renina plasmática, la secreción de aldosterona parece ser exquisitamente sensible incluso a concentraciones subnormales de angiotensina II. Este fenómeno parece ser la base de la eficacia de los BRA en aldosteronismo primario (específicamente hiperplasia suprarrenal idiopática [AHI]).
IRBESARTAN (AVAPRO)
Irbesartan bloquea los efectos vasoconstrictores y secretores de aldosterona de la angiotensina II en el sitio del receptor del tejido. Puede inducir una inhibición más completa del sistema renina-angiotensina que los inhibidores de la ECA. Además, irbesartan no afecta la respuesta a la bradiquinina, y es menos probable que se asocie con tos y angioedema.
LOSARTAN (COZAAR)
Losartan bloquea los efectos vasoconstrictores y secretores de aldosterona de la angiotensina II. Puede inducir una inhibición más completa del sistema renina-angiotensina que los inhibidores de la ECA. Además, losartan no afecta la respuesta a la bradiquinina, y es menos probable que se asocie con tos y angioedema. Es adecuado para pacientes que no pueden tolerar los inhibidores de la ECA.
OLMESARTAN
Olmesartan bloquea los efectos vasoconstrictores de la angiotensina II al bloquear selectivamente la unión de la angiotensina II a los receptores de la angiotensina II tipo 1 en el músculo liso vascular. Su acción es independiente de las vías para la síntesis de angiotensina II.
VALSARTAN (DIOVAN)
Valsartan es un profármaco que desplaza la angiotensina II de los receptores de angiotensina II tipo 1, bloqueando los efectos vasoconstrictores de la angiotensina II. Valsartan también puede disminuir la presión arterial a través de sus efectos sobre la liberación de aldosterona, la liberación de catecolaminas, la liberación de arginina vasopresina, la ingesta de agua y las respuestas hipertróficas.
Valsartan puede inducir una inhibición más completa del sistema renina-angiotensina que los inhibidores de la ECA. Además, no afecta la respuesta a la bradiquinina y es menos probable que se asocie con tos y angioedema. Valsartan es adecuado para pacientes que no pueden tolerar los inhibidores de la ECA.
TELMISARTAN (MICARDIS)
Telmisartan bloquea el vasoconstrictor y los efectos secretores de aldosterona de la angiotensina II al bloquear selectivamente la unión de la angiotensina II al receptor AT1 en muchos tejidos, como el músculo liso vascular y la glándula suprarrenal y, por lo tanto, reduce la presión arterial. También se encuentra un receptor AT2 en muchos tejidos, pero se desconoce si AT2 está asociado con la homeostasis cardiovascular y telmisartan tiene una afinidad mucho mayor por el receptor AT1 que por el receptor AT2.
CORTICOSTEROIDES
Esta clase de agentes son terapéuticamente apropiados solo para el subtipo GRA de aldosteronismo primario. El tratamiento de elección en GRA es la administración de la dosis más baja posible de glucocorticoides que se puede usar para lograr un control adecuado de la presión arterial. Debido a los posibles efectos adversos que pueden resultar incluso de un exceso sutil de glucocorticoides, el uso de glucocorticoides de acción corta, como la prednisona y la hidrocortisona (en lugar de la dexametasona), generalmente es lo mejor.
PREDNISONA
La prednisona es un profármaco de acción corta que ejerce sus efectos después de que se metaboliza y se convierte en prednisolona. La prednisona imita el cortisol de origen natural y se usa en GRA para suprimir rápidamente los niveles de aldosterona y resolver la expansión del volumen y la hipertensión en este trastorno, siendo generalmente eficaz en las 2 semanas posteriores al inicio del tratamiento.
HIDROCORTISONA
La hidrocortisona posee una similitud molecular con la aldosterona y, por lo tanto, no solo se une al receptor de glucocorticoides (resultando en menores niveles de aldosterona en la GRA), sino también al receptor mineralocorticoide (MR), lo que también proporciona una inhibición competitiva de los niveles ambientales de aldosterona en la MR nivel (que resulta en la disminución de los efectos fisiológicos de los niveles ambientales existentes de aldosterona en GRA).
Rossi GP, Ragazzo F, Seccia TM, Maniero C, Barisa M, Calò LA, et al. Hyperparathyroidism can be useful in the identification of primary aldosteronism due to aldosterone-producing adenoma. Hypertension. 2012 Aug. 60(2):431-6. [Medline].
Dluhy RG, Lifton RP. Glucocorticoid-remediable aldosteronism. Endocrinol Metab Clin North Am. 1994 Jun. 23(2):285-97. [Medline].
Funder JW. The genetic basis of primary aldosteronism. Curr Hypertens Rep. 2012 Apr. 14(2):120-4. [Medline].
Choi M, Scholl UI, Yue P, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011 Feb 11. 331(6018):768-72. [Medline]. [Full Text].
Young DB. Quantitative analysis of aldosterone's role in potassium regulation. Am J Physiol. 1988 Nov. 255(5 Pt 2):F811-22. [Medline].
Stowasser M, Klemm SA, Tunny TJ, et al. Plasma aldosterone response to ACTH in subtypes of primary aldosteronism. Clin Exp Pharmacol Physiol. 1995 Jun-Jul. 22(6-7):460-2. [Medline].
Litchfield WR, New MI, Coolidge C, et al. Evaluation of the dexamethasone suppression test for the diagnosis of glucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab. 1997 Nov. 82(11):3570-3. [Medline]. [Full Text].
Mulatero P, Stowasser M, Loh KC, Fardella CE, Gordon RD, Mosso L, et al. Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. J Clin Endocrinol Metab. 2004 Mar. 89(3):1045-50. [Medline].
Born-Frontsberg E, Reincke M, Rump LC, et al. Cardiovascular and cerebrovascular comorbidities of hypokalemic and normokalemic primary aldosteronism: results of the German Conn's Registry. J Clin Endocrinol Metab. 2009 Apr. 94(4):1125-30. [Medline].
Bernini G, Galetta F, Franzoni F, Bardini M, Taurino C, Bernardini M, et al. Arterial stiffness, intima-media thickness and carotid artery fibrosis in patients with primary aldosteronism. J Hypertens. 2008 Dec. 26(12):2399-405. [Medline].
Apostolopoulou K, Künzel HE, Gerum S, Merkle K, Schulz S, Fischer E, et al. Gender differences in anxiety and depressive symptoms in patients with primary hyperaldosteronism: A cross-sectional study. World J Biol Psychiatry. 2012 May 8. [Medline].
Cruz DN, Perazella MA. Hypertension and hypokalemia: unusual syndromes. Conn Med. 1997 Feb. 61(2):67-75. [Medline].
Cesari M, Letizia C, Angeli P, Sciomer S, Rosi S, Rossi GP. Cardiac Remodeling in Patients With Primary and Secondary Aldosteronism: A Tissue Doppler Study. Circ Cardiovasc Imaging. 2016 Jun. 9 (6):[Medline].
Hall JE, Granger JP, Smith MJ Jr. Role of renal hemodynamics and arterial pressure in aldosterone "escape". Hypertension. 1984. 6:1183.
Yokota N, Bruneau BG, Kuroski de Bold ML, et al. Atrial natriuretic factor significantly contributes to the mineralocorticoid escape phenomenon. Evidence for a guanylate cyclase-mediated pathway. J Clin Invest. 1994 Nov. 94(5):1938-46. [Medline]. [Full Text].
Vallon V, Rieg T. Regulation of renal NaCl and water transport by the ATP/UTP/P2Y2 receptor system. Am J Physiol Renal Physiol. 2011 Sep. 301(3):F463-75. [Medline]. [Full Text].
Schiffrin EL, Chrétien M, Seidah NG, et al. Response of human aldosteronoma cells in culture to the N-terminal glycopeptide of pro-opiomelanocortin and gamma 3-MSH. Horm Metab Res. 1983 Apr. 15(4):181-4. [Medline].
[Guideline] Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M, et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2008 Sep. 93(9):3266-81. [Medline].
Quamme GA. Control of magnesium transport in the thick ascending limb. Am J Physiol. 1989 Feb. 256(2 Pt 2):F197-210. [Medline].
Seiler L, Rump LC, Schulte-Mönting J, Slawik M, Borm K, Pavenstädt H, et al. Diagnosis of primary aldosteronism: value of different screening parameters and influence of antihypertensive medication. Eur J Endocrinol. 2004 Mar. 150(3):329-37. [Medline].
Tzanela M, Effremidis G, Vassiliadi D, et al. The aldosterone to renin ratio in the evaluation of patients with incidentally detected adrenal masses. Endocrine. 2007 Oct. 32(2):136-42. [Medline].
Diederich S, Mai K, Bahr V, et al. The simultaneous measurement of plasma-aldosterone- and -renin-concentration allows rapid classification of all disorders of the renin-aldosterone system. Exp Clin Endocrinol Diabetes. 2007 Jul. 115(7):433-8. [Medline].
Wu VC, Chang HW, Liu KL, et al. Primary aldosteronism: diagnostic accuracy of the losartan and captopril tests. Am J Hypertens. 2009 Aug. 22(8):821-7. [Medline].
Burton TJ, Mackenzie IS, Balan K, Koo B, Bird N, Soloviev DV, et al. Evaluation of the sensitivity and specificity of (11)C-metomidate positron emission tomography (PET)-CT for lateralizing aldosterone secretion by Conn's adenomas. J Clin Endocrinol Metab. 2012 Jan. 97(1):100-9. [Medline].
Otsuka F, Otsuka-Misunaga F, Koyama S, et al. Hormonal characteristics of primary aldosteronism due to unilateral adrenal hyperplasia. J Endocrinol Invest. 1998 Sep. 21(8):531-6. [Medline].
Stowasser M, Bachmann AW, Tunny TJ. Production of 18-oxo-cortisol in subtypes of primary aldosteronism. Clin Exp Pharmacol Physiol. 1996 Jun-Jul. 23(6-7):591-3. [Medline].
Hamlet SM, Gordon RD, Gomez-Sanchez CE, et al. Adrenal transitional zone steroids, 18-oxo and 18-hydroxycortisol, useful in the diagnosis of primary aldosteronism, are ACTH-dependent. Clin Exp Pharmacol Physiol. 1988 Apr. 15(4):317-22. [Medline].
McAlister FA, Lewanczuk RZ. Primary hyperaldosteronism and adrenal incidentaloma: an argument for physiologic testing before adrenalectomy. Can J Surg. 1998 Aug. 41(4):299-305. [Medline].
Di Martino M, Garcia Sanz I, Munoz de Nova JL, Marin Campos C, Martinez Martin M, Dominguez Gadea L. NP-59 test for preoperative localization of primary hyperaldosteronism. Langenbecks Arch Surg. 2017 Feb 21. [Medline].
Georgiades CS, Hong K, Geschwind JF, et al. Adjunctive use of C-arm CT may eliminate technical failure in adrenal vein sampling. J Vasc Interv Radiol. 2007 Sep. 18(9):1102-5. [Medline].
Young WF, Stanson AW, Grant CS, et al. Primary aldosteronism: adrenal venous sampling. Surgery. 1996 Dec. 120(6):913-9; discussion 919-20. [Medline].
Webb R, Mathur A, Chang R, Baid S, Nilubol N, Libutti SK, et al. What is the Best Criterion for the Interpretation of Adrenal Vein Sample Results in Patients with Primary Hyperaldosteronism?. Ann Surg Oncol. 2011 Nov 3. [Medline].
Dekkers T, Prejbisz A, Kool LJ, et al. Adrenal vein sampling versus CT scan to determine treatment in primary aldosteronism: an outcome-based randomised diagnostic trial. Lancet Diabetes Endocrinol. 2016 Sep. 4 (9):739-46. [Medline].
Young WF, Stanson AW, Thompson GB, et al. Role for adrenal venous sampling in primary aldosteronism. Surgery. 2004 Dec. 136(6):1227-35. [Medline].
Naruse M, Naruse K, Yoshimoto T, et al. Dopaminergic regulation of aldosterone secretion: its pathophysiologic significance in subsets of primary aldosteronism. Hypertens Res. 1995 Jun. 18 Suppl 1:S59-64. [Medline].
Carey RM. Primary aldosteronism. Horm Res. 2009 Jan. 71 Suppl 1:8-12. [Medline].
Hood SJ, Taylor KP, Ashby MJ, et al. The spironolactone, amiloride, losartan, and thiazide (SALT) double-blind crossover trial in patients with low-renin hypertension and elevated aldosterone-renin ratio. Circulation. 2007 Jul 17. 116(3):268-75. [Medline]. [Full Text].
Ronconi V, Turchi F, Appolloni G, di Tizio V, Boscaro M, Giacchetti G. Aldosterone, Mineralocorticoid Receptor and the Metabolic Syndrome: Role of the Mineralocorticoid Receptor Antagonists. Curr Vasc Pharmacol. 2011 Oct 21. [Medline].
Minowada S, Fujimura T, Takahashi N, et al. Computed tomography-guided percutaneous acetic acid injection therapy for functioning adrenocortical adenoma. J Clin Endocrinol Metab. 2003 Dec. 88(12):5814-7. [Medline]. [Full Text].
Sukor N, Gordon RD, Ku YK, et al. Role of unilateral adrenalectomy in bilateral primary aldosteronism: a 22-year single center experience. J Clin Endocrinol Metab. 2009 Jul. 94(7):2437-45. [Medline].
Celen O, O'Brien MJ, Melby JC, et al. Factors influencing outcome of surgery for primary aldosteronism. Arch Surg. 1996 Jun. 131(6):646-50. [Medline].
Waldmann J, Maurer L, Holler J, Kann PH, Ramaswamy A, Bartsch DK, et al. Outcome of surgery for primary hyperaldosteronism. World J Surg. 2011 Nov. 35(11):2422-7. [Medline].
Letavernier E, Peyrard S, Amar L, et al. Blood pressure outcome of adrenalectomy in patients with primary hyperaldosteronism with or without unilateral adenoma. J Hypertens. 2008 Sep. 26(9):1816-23. [Medline].
Milsom SR, Espiner EA, Nicholls MG, et al. The blood pressure response to unilateral adrenalectomy in primary aldosteronism. Q J Med. 1986 Dec. 61(236):1141-51. [Medline].
Giacchetti G, Turchi F, Boscaro M, Ronconi V. Management of primary aldosteronism: its complications and their outcomes after treatment. Curr Vasc Pharmacol. 2009 Apr. 7(2):244-49. [Medline].
Amar L, Baguet JP, Bardet S, et al. SFE/SFHTA/AFCE primary aldosteronism consensus: Introduction and handbook. Ann Endocrinol (Paris). 2016 Jun 14. [Medline]. [Full Text].
Baguet JP, Steichen O, Mounier-Vehier C, Gosse P. SFE/SFHTA/AFCE consensus on primary aldosteronism, part 1: Epidemiology of PA, who should be screened for sporadic PA?. Ann Endocrinol (Paris). 2016 Apr 15. 86 (20):1002-8. [Medline]. [Full Text].
Douillard C, Houillier P, Nussberger J, Girerd X. SFE/SFHTA/AFCE Consensus on Primary Aldosteronism, part 2: First diagnostic steps. Ann Endocrinol (Paris). 2016 May 10. [Medline]. [Full Text].
Reznik Y, Amar L, Tabarin A. SFE/SFHTA/AFCE consensus on primary aldosteronism, part 3: Confirmatory testing. Ann Endocrinol (Paris). 2016 Jun 15. [Medline]. [Full Text].
Bardet S, Chamontin B, Douillard C, et al. SFE/SFHTA/AFCE consensus on primary aldosteronism, part 4: Subtype diagnosis. Ann Endocrinol (Paris). 2016 Mar 29. [Medline]. [Full Text].
Zennaro MC, Jeunemaitre X. SFE/SFHTA/AFCE consensus on primary aldosteronism, part 5: Genetic diagnosis of primary aldosteronism. Ann Endocrinol (Paris). 2016 Jun 14. [Medline]. [Full Text].
Steichen O, Amar L, Chaffanjon P, Kraimps JL, Menegaux F, Zinzindohoue F. SFE/SFHTA/AFCE consensus on primary aldosteronism, part 6: Adrenal surgery. Ann Endocrinol (Paris). 2016 Jun 10. [Medline]. [Full Text].
Pechère-Bertschi A, Herpin D, Lefebvre H. SFE/SFHTA/AFCE consensus on primary aldosteronism, part 7: Medical treatment of primary aldosteronism. Ann Endocrinol (Paris). 2016 Jun 14. [Medline]. [Full Text].
[Guideline] Funder JW, Carey RM, Mantero F, et al. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016 May. 101 (5):1889-916. [Medline]. [Full Text].
[Guideline] Romero DG, Yanes Cardozo LL. Clinical Practice Guideline for Management of Primary Aldosteronism: What is New in the 2016 Update?. Int J Endocrinol Metab Disord. 2016. 2 (3):[Medline]. [Full Text].