Paciente de 30 años cuadro de evolucion de 1 año que debuta como nódulo indoloro en region de occipital, crece paulatinamente hasta formar esta lesión.



Diagnóstico: Acné queloide nucal
Presentó
Dr. Andrés Alvarado Quezada
Medico Interno de Pregrado en Hospital Vicente Corral Moscoso
Medical Doctor (MD) en Ministerio de Salud Publica del Ecuador
Estudió Medicina en Universidad Estatal de Cuenca
ACNÉ QUELOIDEO / FOLICULITIS ESCLEROSANTE DE LA NUCA (ACNE KELOIDALIS NUCHAE)
INTRODUCCIÓN
El acné queloide nucal (AQN) es un trastorno crónico común que involucra la inflamación y la cicatrización de los folículos pilosos con el posterior desarrollo de pápulas y placas de tipo queloide y alopecia cicatrizal. El sitio característico de afectación es el cuero cabelludo posterior y el cuello. Con poca frecuencia, otras áreas del cuero cabelludo se ven afectadas.
El acné queloide nucal (AQN) es un trastorno crónico común que involucra la inflamación y la cicatrización de los folículos pilosos con el posterior desarrollo de pápulas y placas de tipo queloide y alopecia cicatrizal. El sitio característico de afectación es el cuero cabelludo posterior y el cuello. Con poca frecuencia, otras áreas del cuero cabelludo se ven afectadas.
El AQN típicamente ocurre en hombres de ascendencia africana, pero también se desarrolla ocasionalmente en mujeres y en grupos étnicos no africanos. Puede ser pruriginoso, doloroso o desfigurar cosméticamente, lo que contribuye a los efectos negativos sobre la calidad de vida.
TERMINOLOGÍA
Aunque el término "acné keloidalis nuchae" se usa comúnmente, hay otras descripciones históricas y contemporáneas para AQN. Los términos históricos para esta condición incluyen "sycosis framboesiformis" y "dermatitis papillaris capillitii" [ 1,2 ]. Los nombres contemporáneos alternativos para AQN incluyen "folliculitis keloidalis" [ 3 ], "foliculitis nuchae" [ 4 ] y "foliculitis keloidalis nuchae" [ 5 ]. Además, debido a la extensión ocasional a otras áreas del cuero cabelludo, algunos autores eliminan "nucha" de AQN y se refieren al trastorno como "acné keloidalis".
Aunque el término "acné keloidalis nuchae" se usa comúnmente, hay otras descripciones históricas y contemporáneas para AQN. Los términos históricos para esta condición incluyen "sycosis framboesiformis" y "dermatitis papillaris capillitii" [ 1,2 ]. Los nombres contemporáneos alternativos para AQN incluyen "folliculitis keloidalis" [ 3 ], "foliculitis nuchae" [ 4 ] y "foliculitis keloidalis nuchae" [ 5 ]. Además, debido a la extensión ocasional a otras áreas del cuero cabelludo, algunos autores eliminan "nucha" de AQN y se refieren al trastorno como "acné keloidalis".
Múltiples autores han sugerido que el nombre "acné queloidalis nuchae" es un nombre inapropiado, ya que las lesiones no son patológicamente ni acnéicas ni queloides [ 6 ]. En contraste con el acné verdadero, los comedones nunca están presentes en la AQN [ 7 ]. Las lesiones parecidas a cicatrices, aunque se asemejan a queloides, no suelen reaparecer después de la escisión, como lo hacen los queloides a menudo [ 8 ], y los pacientes generalmente no tienen queloides en ningún otro lugar.
EPIDEMIOLOGÍA
AQN es un trastorno común. Las personas de ascendencia africana con cabello con textura Afro son la población predominante en riesgo. Esta predilección fue evidente en un estudio de 453 jugadores de fútbol masculino (de 14 a 27 años); AQN estuvo presente en el 14 por ciento de los jugadores negros en comparación con el 0 por ciento de los jugadores blancos [ 9 ]. Además, los estudios realizados en América del Norte, el Caribe y África han encontrado que AQN representa entre el 0,45 y el 9 por ciento de los diagnósticos entre pacientes negros evaluados en clínicas de dermatología [ 6,10-14 ].
AQN es un trastorno común. Las personas de ascendencia africana con cabello con textura Afro son la población predominante en riesgo. Esta predilección fue evidente en un estudio de 453 jugadores de fútbol masculino (de 14 a 27 años); AQN estuvo presente en el 14 por ciento de los jugadores negros en comparación con el 0 por ciento de los jugadores blancos [ 9 ]. Además, los estudios realizados en América del Norte, el Caribe y África han encontrado que AQN representa entre el 0,45 y el 9 por ciento de los diagnósticos entre pacientes negros evaluados en clínicas de dermatología [ 6,10-14 ].
AQN afecta a los hombres con mucha más frecuencia que las mujeres, aunque las mujeres ocasionalmente desarrollan la desarrollan [ 15,16 ]. Una proporción de 20: 1 de hombres afectados a mujeres se cita comúnmente [ 17 ]. En un grupo seleccionado por la comunidad en Sudáfrica, el 11 por ciento de 267 varones versus el 0.3 por ciento de las 597 mujeres tenían AQN [ 18 ].
El inicio de la enfermedad generalmente sigue a la adolescencia y es raro después de los 50 [ 19 ]. La predilección por la aparición de AQN después de la adolescencia se demostró en un estudio de 1042 escolares sudafricanos (de 6 a 21 años de edad). La prevalencia de AQN fue del 0,67 por ciento en general, pero entre los estudiantes en el último año de la escuela secundaria, la prevalencia fue del 4,7 por ciento [ 18 ].
PATOGENIA
Como se evidencia por una multitud de teorías propuestas con niveles variables de evidencia, la causa de AQN no se ha establecido definitivamente. Mecanismos patógenos relacionados con la lesión cutánea y respuestas inmunes aberrantes a antígenos comunes se encuentran entre las teorías citadas.
Como se evidencia por una multitud de teorías propuestas con niveles variables de evidencia, la causa de AQN no se ha establecido definitivamente. Mecanismos patógenos relacionados con la lesión cutánea y respuestas inmunes aberrantes a antígenos comunes se encuentran entre las teorías citadas.
● Lesión en la piel : algunas teorías de causalidad se centran en la irritación crónica u oclusión de los folículos debido a prácticas de corte de pelo (por ejemplo, rasurado o estrechamiento), traumas, fricción (por ejemplo, frotamiento de cuellos de camisa o cascos), calor o humedad como un factor que predispone o exacerba [ 11,20-23 ]. No está claro si la herramienta utilizada para cortar el cabello podría influir en el riesgo de AQN; en un estudio sudafricano, la prevalencia de AQN no pareció diferir con el uso de afeitadoras versus cortaúñas [ 18 ]. Los cortes de pelo frecuentes y traumáticos también se han propuesto como potenciales contribuyentes, aunque es necesario realizar más estudios para determinar si estos son factores relevantes [ 10,18 ].
Es poco probable que los insultos externos sean la única causa de AQN, dado el número desproporcionado de casos que ocurren en hombres afrodescendientes. Los hallazgos de un estudio de 453 jugadores de fútbol masculino respaldan esto; en contraste con la mayor prevalencia de AQN observada en jugadores negros, el acné mecánico (una erupción acneiforme inducida por fricción, oclusión y calor [ 24 ]) se produjo en la parte posterior del cuello a tasas similares en jugadores de blancos y negros [ 9 ]. Se ha sugerido un papel para el tipo de cabello afro-texturizado en AQN en base a la mayor prevalencia en la población negra, posiblemente relacionada con la curvatura y la morfología del cabello con textura afro. Aunque la penetración del cabello curvo en la piel se ha propuesto como un mecanismo potencial [ 17], esta teoría ha sido cuestionada por autores que no han encontrado evidencia clínica o patológica de pelos curvándose y reingresando a la piel [ 25,26 ].
● Reacción inmunitaria aberrante : otra teoría patogénica propuesta se basa en la consideración de AQN como una forma de alopecia cicatrizal primaria estimulada por una respuesta inmune en la piel [ 27 ].
La siguiente secuencia de eventos para el desarrollo de AQN como una alopecia cicatricial primaria se ha propuesto [ 27 ]:
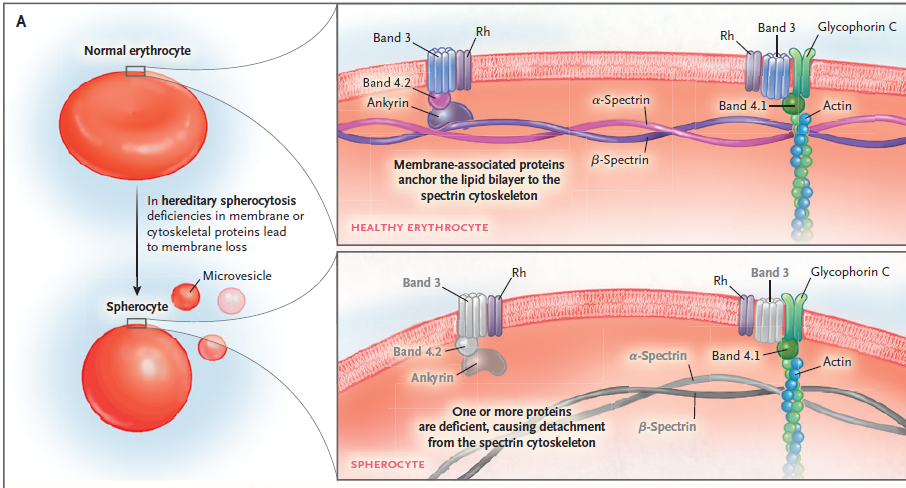
• Los antígenos intrafoliculares (particularmente aquellos que son más prevalentes después de la pubertad) atraen las células inflamatorias al folículo a nivel del istmo ( esquema ). Los antígenos propuestos incluyen Demodex , flora cutánea (por ejemplo, bacterias o esporas de hongos), cosméticos, sebo y queratinocitos descamados.
• La glándula sebácea es un blanco temprano para la inflamación o se destruye de manera secundaria como resultado del proceso inflamatorio.
• La pared folicular se debilita por la inflamación perifolicular, lo que produce espongiosis y una leve exocitosis linfocítica.
• La pared folicular debilitada permite una mayor fuga de antígenos, lo que resulta en una mayor inflamación.
• El proceso continúa con el desarrollo de la fibroplasia lamelar concéntrica, con el pelo penetrando finalmente en la pared folicular y entrando en la dermis, lo que resulta en una intensa inflamación y destrucción epitelial, además, resulta en una cicatriz folicular.
• La cicatrización hipertrófica se desarrolla debido a la presencia de fragmentos del eje del pelo y componentes epiteliales degenerativos en la dermis.
● Otros : otros posibles factores contributivos propuestos basados en datos limitados incluyen autoinmunidad [ 25 ], exceso de andrógenos o aumento de la sensibilidad a los andrógenos [ 13,28 ], seborrea [ 13 ] y medicamentos (p. Ej., Ciclosporina [ 29-31 ], tacrolimus [ 32 ], sirolimus [ 32 ] o difenilhidantoína y carbamazepina [ 33]]). Al igual que con otros colaboradores propuestos, un papel para estos factores aún no se ha confirmado. AQN también se ha reportado en dos pacientes con queratosis folicular de spinulosa decalvans, un trastorno genético ligado a X que predispone a los pacientes al desarrollo de hiperqueratosis e inflamación folicular [ 34,35 ].

Esquema : Anatomía del folículo piloso
MANIFESTACIONES CLÍNICAS
El AQN se presenta con grados variables de inflamación y cicatrización queloidea en el cuero cabelludo inferior posterior y en la nuca. Ocasionalmente, la afectación se extiende al vértice u otras áreas del cuero cabelludo ( figura 1 ) [ 36 ].
El AQN se presenta con grados variables de inflamación y cicatrización queloidea en el cuero cabelludo inferior posterior y en la nuca. Ocasionalmente, la afectación se extiende al vértice u otras áreas del cuero cabelludo ( figura 1 ) [ 36 ].

Figura 1 Alopecia cicatricial en un paciente con acné qeloide nucal. Ocasionalmente, el acné keloidalis nuchae se extiende más allá de la parte superior posterior del cuello y del cuero cabelludo posterior.
La inflamación activa en AQN se manifiesta como pápulas o pústulas inflamadas ( figura 2A-G).
![]()
Acné queloide leve. Múltiples pápulas foliculares en la región posterior del cuero cabelludo y la parte alta del cuello.
![]()
Pápulas pequeñas e inflamadas en le región de la nuca
![]()
Coalescencia de pápulas queloides en la parte superior de la imagen.
![]()
Múltiples pápulas inflamadas coalescentes en lanuca y porción superior del cuello
![]()
Grandes placas y pústulas en la región de la nuca
![]()
Pápulas queloides foliculares inflamadas y pústulas secas en la parte superior de la imagen
![]() Cicatrices, pápulas y pústulas foliculares inflamadas
Cicatrices, pápulas y pústulas foliculares inflamadas
![]()
Alopecia y múltiples placas queloide like coalesciendo para formar nódulos y placas.
![]()
![]()
![]()
![]()
Múltiple placas queloides like con forma de domo
La inflamación puede ser aguda, y puede mostrar eritema marcado, pápulas inflamadas o papulopústulas. Alternativamente, puede estar presente una combinación de inflamación aguda e inflamación crónica (que se manifiesta como pápulas de color rosado a rojo oscuro sin pústulas o secreciones). El grado de inflamación varía de leve a grave y puede variar hasta cierto punto entre los episodios de brote en un individuo dado. Encostramiento o excoriación pueden ser evidentes. En casos severos, puede haber estenosis drenantes o supuración.

Acné queloide leve. Múltiples pápulas foliculares en la región posterior del cuero cabelludo y la parte alta del cuello.

Pápulas pequeñas e inflamadas en le región de la nuca

Coalescencia de pápulas queloides en la parte superior de la imagen.

Múltiples pápulas inflamadas coalescentes en lanuca y porción superior del cuello

Grandes placas y pústulas en la región de la nuca

Pápulas queloides foliculares inflamadas y pústulas secas en la parte superior de la imagen


Alopecia y múltiples placas queloide like coalesciendo para formar nódulos y placas.

Placa cicatrizal en la región posterior del cuero cabelludo

Placa grande cicatrizal

Placa cicatrizal con nódulos queloides

Múltiple placas queloides like con forma de domo
La inflamación puede ser aguda, y puede mostrar eritema marcado, pápulas inflamadas o papulopústulas. Alternativamente, puede estar presente una combinación de inflamación aguda e inflamación crónica (que se manifiesta como pápulas de color rosado a rojo oscuro sin pústulas o secreciones). El grado de inflamación varía de leve a grave y puede variar hasta cierto punto entre los episodios de brote en un individuo dado. Encostramiento o excoriación pueden ser evidentes. En casos severos, puede haber estenosis drenantes o supuración.
La cicatrización generalmente está presente cuando los pacientes se presentan para la evaluación médica. La cicatrización se manifiesta por pápulas, placas o nódulos queloides de tamaño variable en el sitio de los folículos pilosos previamente involucrados. Las pápulas queloides de AQN son típicamente en forma de cúpula. En las etapas posteriores de AQN, los pacientes pueden presentar cicatrización crónica o alopecia cicatricial sin inflamación activa. Pueden aparecer "pelos de muñeca", múltiples pelos que salen de un solo orificio folicular.
AQN puede ser asintomático o sintomático. El prurito o el dolor pueden acompañar a las lesiones de la piel, particularmente si están presentes las pústulas [ 6,17,20,25 ].
CURSO CLÍNICO
AQN es una enfermedad crónica con brotes episódicos de inflamación que empeora. El tratamiento efectivo generalmente requiere un régimen de mantenimiento con ajustes terapéuticos según sea necesario para las erupciones. Con el tiempo (generalmente durante un número variable de años), la enfermedad tiende a ser menos activa en términos de inflamación. Sin embargo, las pápulas y las placas queloides generalmente persisten a menos que se traten.
AQN es una enfermedad crónica con brotes episódicos de inflamación que empeora. El tratamiento efectivo generalmente requiere un régimen de mantenimiento con ajustes terapéuticos según sea necesario para las erupciones. Con el tiempo (generalmente durante un número variable de años), la enfermedad tiende a ser menos activa en términos de inflamación. Sin embargo, las pápulas y las placas queloides generalmente persisten a menos que se traten.
HISTOPATOLOGÍA
Los hallazgos histopatológicos varían según la etapa de la enfermedad y las características clínicas específicas presentes en el área inmediata de la biopsia. Típicamente, sin embargo, AQN se caracteriza por la presencia de foliculitis aguda y crónica, unidades pilosebáceas rotas y fibrosis dérmica densa (tipo cicatricial) ( figura 4A-E ).
![]()
Figura A: Fibrosis dérmica densa e inflamación de la unidad pilosebácea.
Los hallazgos histopatológicos varían según la etapa de la enfermedad y las características clínicas específicas presentes en el área inmediata de la biopsia. Típicamente, sin embargo, AQN se caracteriza por la presencia de foliculitis aguda y crónica, unidades pilosebáceas rotas y fibrosis dérmica densa (tipo cicatricial) ( figura 4A-E ).

Figura A: Fibrosis dérmica densa e inflamación de la unidad pilosebácea.

Figura B: Inflamación que involucra el folículo piloso, pelo extruído por células inflamatorias y fibrosis dérmica

Figura C: Ruptura de la unidad pilosebáceaasociada a microabscesos. Cuatro aumentos en A y diez aumentos en B)

Figura D: Pelo extruído en dermis con infiltrado predominantemente neutrofílicosiguiendo a la destrucción del folículopiloso

Figura E: Intensa fibrosis dérmica
● Etapas iniciales : infiltrado inflamatorio denso folicular y perifolicular de linfocitos y neutrófilos. La fibrosis dérmica se parece más a las fibras de colágeno que se ven en el tejido cicatricial típico que a la fibrosis que se observa en las cicatrices queloides [ 17 ].
● Etapas posteriores : los folículos pilosos rotos y rotos están rodeados de inflamación granulomatosa, formación de abscesos perifoliculares y fibrosis dérmica [ 37 ].
DIAGNÓSTICO
El diagnóstico de AQN generalmente se puede hacer según los hallazgos clínicos. La biopsia rara vez es necesaria. Los hallazgos clínicos que despiertan la sospecha de un diagnóstico de AQN son pápulas o placas queloides firmes en el cuero cabelludo posterior inferior o el cuello posterior superior, con o sin pápulas foliculares inflamadas o papulopustulas.
El diagnóstico de AQN generalmente se puede hacer según los hallazgos clínicos. La biopsia rara vez es necesaria. Los hallazgos clínicos que despiertan la sospecha de un diagnóstico de AQN son pápulas o placas queloides firmes en el cuero cabelludo posterior inferior o el cuello posterior superior, con o sin pápulas foliculares inflamadas o papulopustulas.
● Historial del paciente: el historial del paciente permite al clínico recopilar información que puede ayudar a respaldar o negar el diagnóstico de AQN, determinar el mejor enfoque para el tratamiento y evaluar la respuesta al tratamiento. El conocimiento de lo siguiente es útil:
• Edad de inicio (después de la adolescencia hasta la mediana edad es típico de AQN)
• Duración de la enfermedad y tasa de progresión
• Síntomas (p. Ej., Dolor, prurito, drenaje)
• Prácticas de cuidado del cabello (corte de cabello y afeitado)
• factores exacerbadores
• Tratamientos previos (incluida la respuesta a cada tratamiento)
EXAMEN FÍSICO
Se debe examinar todo el cuero cabelludo. En el típico acné keloidalis nuchae, el área de la enfermedad (pápulas inflamatorias, pústulas y pápulas o placas queloides) se limita al cuero cabelludo y la nuca posteriores inferiores. Encostramiento, excoriación, y en casos severos, supuración, puede estar presente.
Se debe examinar todo el cuero cabelludo. En el típico acné keloidalis nuchae, el área de la enfermedad (pápulas inflamatorias, pústulas y pápulas o placas queloides) se limita al cuero cabelludo y la nuca posteriores inferiores. Encostramiento, excoriación, y en casos severos, supuración, puede estar presente.
Aunque AQN ocasionalmente se extiende a otras áreas del cuero cabelludo [ 36 ], los hallazgos de pápulas foliculares, inflamación, supuración o cicatrización en otras áreas del cuero cabelludo deben provocar la consideración de diagnósticos alternativos, como disección de la celulitis del cuero cabelludo, liquen planopilaris, folículo linfoma cutáneo de células T, tinea capitis u otro trastorno.
● BIOPSIA : la biopsia generalmente es innecesaria a menos que la presentación sea atípica. Si es necesaria una biopsia para el diagnóstico, se debe realizar una biopsia con punción de 3 a 4 mm. El corte vertical de la muestra de patología suele ser suficiente para reconocer las características patológicas de AQN. Si la presentación es atípica o si hay características superpuestas que sugieren un diagnóstico alternativo, se pueden realizar dos biopsias para minimizar el riesgo de diagnóstico erróneo debido a un error de muestreo.
Es esencial que la biopsia sea de suficiente profundidad para incluir la base del folículo. También es importante incluir una sospecha de pápula queloide. La biopsia únicamente de áreas de inflamación severa o supuración franca en ausencia de pápulas queloides puede no ser útil.
● Otras pruebas : si hay posibilidad de sobreinfección (eritema extenso, supuración, dolor, drenaje, olor o síntomas sistémicos como fiebre), se debe obtener un cultivo bacteriano. La necesidad de otras pruebas está determinada por el diagnóstico diferencial. Por ejemplo, si se sospecha tinea capitis, está indicada una preparación de hidróxido de potasio (KOH) o cultivo fúngico.
DIAGNÓSTICO DIFERENCIAL
El diagnóstico de AQN suele ser sencillo, según el aspecto clínico y la ubicación de las lesiones. Sin embargo, hay varios trastornos que pueden merecer consideración si se enfrentan a una presentación atípica de AKN.
El diagnóstico de AQN suele ser sencillo, según el aspecto clínico y la ubicación de las lesiones. Sin embargo, hay varios trastornos que pueden merecer consideración si se enfrentan a una presentación atípica de AKN.
● Acné mecánico
● Foliculitis bacteriana:
● Molusco contagioso
● Foliculitis decalvans
● Celulitis disecante del cuero cabelludo
● Liquen planopilaris
● Linfoma cutáneo de células T (LCCT)
● Tiña de la cabeza
MANEJO
Los datos de tratamiento sobre AQN son limitados y no hay pautas de consenso con respecto a la terapia [ 40 ]. Las decisiones de tratamiento generalmente se guían por la experiencia del médico y se modifican en función de la presentación clínica y las preferencias del paciente. La falta de consenso es evidente en las sugerencias de tratamiento ampliamente variables hechas por un panel de dermatólogos en una revisión de AQN [ 4 ].
Los datos de tratamiento sobre AQN son limitados y no hay pautas de consenso con respecto a la terapia [ 40 ]. Las decisiones de tratamiento generalmente se guían por la experiencia del médico y se modifican en función de la presentación clínica y las preferencias del paciente. La falta de consenso es evidente en las sugerencias de tratamiento ampliamente variables hechas por un panel de dermatólogos en una revisión de AQN [ 4 ].
APROXIMACIÓN AL TRATAMIENTO
El manejo de la AQN consiste en medidas dirigidas a mejorar las manifestaciones clínicas y los síntomas. Nuestras intervenciones iniciales generalmente incluyen asesoramiento de pacientes para evitar posibles factores exacerbadores y la administración de medicamentos para mejorar la inflamación activa. La reducción al mínimo de la apariencia de las cicatrices se puede abordar simultáneamente o siguiendo el control de la inflamación activa.
El manejo de la AQN consiste en medidas dirigidas a mejorar las manifestaciones clínicas y los síntomas. Nuestras intervenciones iniciales generalmente incluyen asesoramiento de pacientes para evitar posibles factores exacerbadores y la administración de medicamentos para mejorar la inflamación activa. La reducción al mínimo de la apariencia de las cicatrices se puede abordar simultáneamente o siguiendo el control de la inflamación activa.
EVITACIÓN DE FACTORES EXACERBADORES
Aunque la causa de AQN no se conoce definitivamente, se han propuesto una variedad de factores como factores agravantes de la afección. En un intento de minimizar las exacerbaciones de la enfermedad, alentamos a los pacientes a [ 6,17,36,41 ]:
Aunque la causa de AQN no se conoce definitivamente, se han propuesto una variedad de factores como factores agravantes de la afección. En un intento de minimizar las exacerbaciones de la enfermedad, alentamos a los pacientes a [ 6,17,36,41 ]:
● Evite recoger, frotar o arañar el área afectada
● Interrumpir el afeitado, el recorte o la rasurado o afeitado de la línea posterior del cabello
● Evite la irritación causada por sombreros ajustados, cascos o camisas de cuello alto
Sin embargo, la eficacia de tales medidas en AQN no ha sido estudiada.
TRATAMIENTO DE LA INFLAMACIÓN
Los medicamentos tópicos o sistémicos con propiedades antiinflamatorias representan el método principal para mejorar las manifestaciones inflamatorias de AQN (pápulas y pápulas inflamatorias). El tratamiento y la prevención de infecciones secundarias también pueden ayudar a reducir los síntomas y minimizar las exacerbaciones.
Los medicamentos tópicos o sistémicos con propiedades antiinflamatorias representan el método principal para mejorar las manifestaciones inflamatorias de AQN (pápulas y pápulas inflamatorias). El tratamiento y la prevención de infecciones secundarias también pueden ayudar a reducir los síntomas y minimizar las exacerbaciones.
INFLAMACIÓN DE LEVE A MODERADA
Los pacientes que presentan pápulas o pústulas inflamatorias pequeñas a menudo pueden lograr una mejoría en los síntomas con la terapia tópica. Los efectos antiinflamatorios de los corticosteroides tópicos son útiles para este propósito. A menudo combinamos la terapia con corticosteroides tópicos con la terapia antibacteriana tópica o la terapia con retinoides tópicos en un intento de obtener un beneficio adicional.
Los pacientes que presentan pápulas o pústulas inflamatorias pequeñas a menudo pueden lograr una mejoría en los síntomas con la terapia tópica. Los efectos antiinflamatorios de los corticosteroides tópicos son útiles para este propósito. A menudo combinamos la terapia con corticosteroides tópicos con la terapia antibacteriana tópica o la terapia con retinoides tópicos en un intento de obtener un beneficio adicional.
CORTICOSTEROIDES TÓPICOS
A pesar del uso frecuente de corticosteroides tópicos para AQN, existen pocos datos sobre la eficacia. Algunos apoyos provienen de un estudio abierto en el que 20 hombres y mujeres afroamericanos con AQN leve a moderada (definida como menos de 50 pápulas o pústulas) fueron tratados con propionato tópico de clobetasol al 0.05% de espuma (aplicado dos veces al día durante dos semanas alternando sin terapia durante dos semanas) durante un período de ocho semanas [ 42 ]. Al final del tratamiento, el recuento medio de pápulas y pápulas se redujo significativamente en comparación con el valor inicial. Los pacientes que continuaron con la enfermedad activa después de este período fueron tratados con betametasonaValerato 0,12% de espuma (un corticosteroide tópico menos potente) durante cuatro semanas adicionales. Sin embargo, no se observó una diferencia significativa en la respuesta entre 8 y 12 semanas.
A pesar del uso frecuente de corticosteroides tópicos para AQN, existen pocos datos sobre la eficacia. Algunos apoyos provienen de un estudio abierto en el que 20 hombres y mujeres afroamericanos con AQN leve a moderada (definida como menos de 50 pápulas o pústulas) fueron tratados con propionato tópico de clobetasol al 0.05% de espuma (aplicado dos veces al día durante dos semanas alternando sin terapia durante dos semanas) durante un período de ocho semanas [ 42 ]. Al final del tratamiento, el recuento medio de pápulas y pápulas se redujo significativamente en comparación con el valor inicial. Los pacientes que continuaron con la enfermedad activa después de este período fueron tratados con betametasonaValerato 0,12% de espuma (un corticosteroide tópico menos potente) durante cuatro semanas adicionales. Sin embargo, no se observó una diferencia significativa en la respuesta entre 8 y 12 semanas.
Cuando tratamos pacientes con AQN, generalmente prescribimos un corticosteroide tópico de alta potencia (grupo 1 o grupo 2 ) e instruimos al paciente a aplicar el corticosteroide una o dos veces al día en el área afectada. Debido a que generalmente, al mismo tiempo, le recetamos un antibiótico tópico o un retinoide tópico, con mayor frecuencia le pedimos a los pacientes que usen el corticosteroide tópico una vez al día (consulte "Terapias complementarias").abajo). El corticosteroide tópico se aplica antes de acostarse. Con el fin de minimizar el riesgo de efectos secundarios de corticosteroides, instruimos a los pacientes a utilizar el corticosteroide tópico en ciclos de dos semanas que se alternan con intervalos de dos semanas sin tratamiento. Los corticosteroides tópicos están disponibles en una variedad de vehículos (por ejemplo, ungüentos, espumas, geles, cremas, soluciones), que se pueden seleccionar según las preferencias del paciente. Encontramos que muchos de nuestros pacientes prefieren ungüentos o lociones, ya que estas formulaciones tienden a ser más calmantes y cosméticamente aceptables.
Cabe destacar que, en el caso de una supuración severa o muchas pústulas en el cuero cabelludo, lo que sugiere una superinfección, retrasamos el tratamiento tópico con corticosteroides. La terapia con corticosteroides tópicos puede comenzar después del tratamiento con antibióticos orales y la resolución de los síntomas sugestivos de infección o supuración.
Reevaluamos a los pacientes cada cuatro a seis semanas para evaluar la respuesta al tratamiento y observar los efectos adversos. Esperamos una respuesta a la terapia con corticosteroides tópicos (reducción notable de la gravedad de las pápulas y pápulas) dentro de las primeras seis a ocho semanas de tratamiento, seguida de una mejora continua con la terapia continua. Una vez que se logra una mejoría suficiente, descontinuamos el corticosteroide tópico con el plan de reiniciar el tratamiento si los síntomas vuelven a aparecer. Si se observa poca mejoría después de seis a ocho semanas de tratamiento, hacemos la transición a otras terapias.
Los efectos adversos potenciales de los corticosteroides tópicos incluyen atrofia cutánea e hipopigmentación en el sitio de tratamiento. Por lo tanto, se debe advertir a los pacientes que apliquen el corticosteroide solo en las áreas afectadas del cuero cabelludo y que eviten aplicar el corticosteroide durante períodos prolongados sin supervisión médica. Los efectos adversos adicionales de los corticosteroides tópicos se revisan por separado. (Consulte "Principios generales de la terapia dermatológica y el uso tópico de corticosteroides", sección "Efectos secundarios" ).
TERAPIAS ADYUVANTES
Generalmente combinamos la terapia con corticosteroides tópicos con un agente adicional en un intento de aumentar la respuesta al tratamiento. Para pacientes con un componente pustular prominente, generalmente agregamos un antimicrobiano tópico. Para pacientes con pápulas principalmente inflamatorias, generalmente agregamos un retinoide tópico. Aunque los antibacterianos tópicos y los retinoides tópicos son terapias de uso común, la eficacia de estos agentes para AQN no se ha evaluado en estudios de investigación.
Generalmente combinamos la terapia con corticosteroides tópicos con un agente adicional en un intento de aumentar la respuesta al tratamiento. Para pacientes con un componente pustular prominente, generalmente agregamos un antimicrobiano tópico. Para pacientes con pápulas principalmente inflamatorias, generalmente agregamos un retinoide tópico. Aunque los antibacterianos tópicos y los retinoides tópicos son terapias de uso común, la eficacia de estos agentes para AQN no se ha evaluado en estudios de investigación.
AGENTES ANTIBACTERIANOS TÓPICOS
Aunque AQN no es una enfermedad principalmente infecciosa, la experiencia clínica sugiere que los agentes antibacterianos tópicos a veces son útiles para mejorar la AQN. Estos agentes pueden ayudar a minimizar las exacerbaciones de la enfermedad a través del tratamiento y la prevención de la infección secundaria. Los efectos antiinflamatorios de los antibacterianos también pueden ser beneficiosos.
Aunque AQN no es una enfermedad principalmente infecciosa, la experiencia clínica sugiere que los agentes antibacterianos tópicos a veces son útiles para mejorar la AQN. Estos agentes pueden ayudar a minimizar las exacerbaciones de la enfermedad a través del tratamiento y la prevención de la infección secundaria. Los efectos antiinflamatorios de los antibacterianos también pueden ser beneficiosos.
Normalmente, le recetamos una solución tópica de clindamicina , que se aplica en el área afectada una vez al día por la mañana. La aplicación del corticosteroide tópico se puede hacer por la noche. Los productos tópicos alternativos incluyen eritromicina , peróxido de benzoilo , peróxido de benzoilo / gel de combinación de clindamicina o gel de combinación de peróxido de benzoilo / eritromicina . La eficacia comparativa de diferentes agentes antimicrobianos tópicos para AQN es desconocida. Las formulaciones de gel, solución, espuma o aderezo de agentes antibacterianos tópicos pueden ser adicionalmente útiles para sus efectos de secado sobre las pústulas.
Los posibles efectos adversos de los agentes antibacterianos tópicos incluyen reacciones alérgicas o irritantes. Si los geles, las soluciones o las formulaciones de los aderezos son demasiado irritantes, las formulaciones de loción son una alternativa. Los pacientes que usan peróxido de benzoilo deben ser informados del potencial del producto para blanquear la ropa o las sábanas.
RETINOIDES TÓPICOS
Se ha teorizado que los retinoides tópicos pueden ayudar a mejorar la AKN al normalizar la maduración y descamación de los queratinocitos foliculares, lo que disminuye el bloqueo folicular. Los retinoides tópicos también tienen propiedades antiinflamatorias [ 43,44 ].
Se ha teorizado que los retinoides tópicos pueden ayudar a mejorar la AKN al normalizar la maduración y descamación de los queratinocitos foliculares, lo que disminuye el bloqueo folicular. Los retinoides tópicos también tienen propiedades antiinflamatorias [ 43,44 ].
Los retinoides tópicos disponibles para su uso en los Estados Unidos incluyen tretinoína, adapaleno y tazaroteno . La eficacia de estos agentes para AKN no se ha comparado; por lo tanto, la eficacia relativa no está clara.
La irritación es un efecto secundario común de los retinoides tópicos, particularmente al inicio de la terapia. Normalmente utilizamos la crema de tretinoína, comenzando con la concentración más baja (0.025%) aplicada cada dos días, y aumentamos a la aplicación diaria según lo toleramos. La fuerza de la tretinoína también puede aumentar, dependiendo de la respuesta. La tretinoína tópica se debe aplicar por la noche, ya que la droga está sujeta a fotodegradación. Por lo tanto, cuando tratamos a pacientes con tretinoína tópica y un corticosteroide tópico, le pedimos al paciente que aplique el corticosteroide tópico por la mañana. Adapaleney algunas formulaciones especializadas de tretinoína son más estables a la luz que la tretinoína estándar; sin embargo, en la práctica, normalmente aún aplicamos estos por la noche. Si es beneficioso, la terapia con retinoides tópicos puede continuar indefinidamente.
El uso de retinoides tópicos durante el embarazo no se recomienda. En particular, el tazaroteno , considerado uno de los retinoides tópicos más efectivos en el contexto del tratamiento del acné, está contraindicado en el embarazo (categoría X ( tabla 2 )). Los efectos adversos de los retinoides tópicos se revisan con mayor detalle por separado. (Consulte "Tratamiento del acné vulgar", sección sobre "Efectos adversos" ).
INFLAMACIÓN DE MODERADA A GRAVE
Cuando los pacientes presentan numerosas lesiones inflamatorias, pustulación extensa o supuración, o lesiones inflamatorias más grandes, alteramos el enfoque de la terapia. Los antibióticos orales y los corticosteroides intralesionales pueden ser útiles para mejorar los signos y síntomas de AKN en estos pacientes. Estos medicamentos también representan un enfoque de segunda línea para los pacientes con enfermedad más leve que no mejoran con la terapia tópica. En ausencia de infección, generalmente combinamos el tratamiento antibiótico oral con terapia con corticosteroides tópicos o intralesionales.
Cuando los pacientes presentan numerosas lesiones inflamatorias, pustulación extensa o supuración, o lesiones inflamatorias más grandes, alteramos el enfoque de la terapia. Los antibióticos orales y los corticosteroides intralesionales pueden ser útiles para mejorar los signos y síntomas de AKN en estos pacientes. Estos medicamentos también representan un enfoque de segunda línea para los pacientes con enfermedad más leve que no mejoran con la terapia tópica. En ausencia de infección, generalmente combinamos el tratamiento antibiótico oral con terapia con corticosteroides tópicos o intralesionales.
LOS ANTIBIÓTICOS ORALES
Los antibióticos orales se reservan principalmente para los brotes graves de la inflamación. Los derivados de tetraciclina se recetan con mayor frecuencia para AQN debido a su cobertura Gram positiva y efectos antiinflamatorios. Sin embargo, si la superinfección es una preocupación (eritema extenso, supuración, dolor, drenaje, olor o síntomas sistémicos como fiebre), se debe obtener un cultivo bacteriano y sensibilidades, y la terapia con antibióticos inicial debe ajustarse en consecuencia.
Los antibióticos orales se reservan principalmente para los brotes graves de la inflamación. Los derivados de tetraciclina se recetan con mayor frecuencia para AQN debido a su cobertura Gram positiva y efectos antiinflamatorios. Sin embargo, si la superinfección es una preocupación (eritema extenso, supuración, dolor, drenaje, olor o síntomas sistémicos como fiebre), se debe obtener un cultivo bacteriano y sensibilidades, y la terapia con antibióticos inicial debe ajustarse en consecuencia.
Como con la mayoría de los otros tratamientos para AQN, la eficacia de los antibióticos orales no se ha estudiado. El uso de estos agentes se basa en la experiencia clínica que sugiere un beneficio para mejorar la inflamación.
Normalmente tratamos a adultos con AKN con doxiciclina oral (100 mg dos veces al día) durante varias semanas a varios meses, dependiendo de la respuesta al tratamiento. La minociclina (75 a 100 mg dos veces al día) es una alternativa, pero existe un mayor riesgo de eventos adversos. Reevaluamos a los pacientes después de seis a ocho semanas de tratamiento y esperamos ver al menos una respuesta parcial en este período. Una vez que se controla la inflamación, disminuimos la doxiciclina según la tolerancia a la dosis más baja que mantiene la respuesta al tratamiento. El objetivo es interrumpir eventualmente la terapia con antibióticos orales y mantener la mejoría con las terapias tópicas que se usan para la inflamación más leve. (Consulte 'Inflamación leve a moderada' más arriba).
Si aparecen bengalas durante o después de la disminución, le indicamos a los pacientes que reinicien la doxiciclina 100 mg dos veces al día. Otros antibióticos que se han usado para AKN incluyen cefalosporinas, clindamicina y eritromicina .
La doxiciclina y la minociclina son generalmente bien toleradas. Los efectos secundarios comunes de los derivados de tetraciclina incluyen fotosensibilidad y malestar gastrointestinal. Además, la minociclina se ha asociado con vértigo, pigmentación de la piel, enfermedad del suero, reacción farmacológica con eosinofilia y síntomas sistémicos (DRESS), anomalías hepáticas y un síndrome similar al lupus. (Consulte "Tratamiento del acné vulgar", sección sobre "Tetraciclinas" ).
CORTICOSTEROIDES INTRALESIONALES
Como en muchas otras afecciones inflamatorias de la piel, las inyecciones intralesionales de corticosteroides pueden ser útiles para tratar las pápulas inflamatorias persistentes que no se pueden resolver con la terapia tópica. Normalmente inyectamos acetónido de triamcinolona (2.5 a 5 mg / ml) en pápulas inflamadas y reevaluamos en cuatro semanas. La atrofia cutánea y la hipopigmentación se encuentran entre los efectos secundarios más comunes de esta terapia.
Como en muchas otras afecciones inflamatorias de la piel, las inyecciones intralesionales de corticosteroides pueden ser útiles para tratar las pápulas inflamatorias persistentes que no se pueden resolver con la terapia tópica. Normalmente inyectamos acetónido de triamcinolona (2.5 a 5 mg / ml) en pápulas inflamadas y reevaluamos en cuatro semanas. La atrofia cutánea y la hipopigmentación se encuentran entre los efectos secundarios más comunes de esta terapia.
La inyección intralesional de corticosteroides también puede ser útil para suavizar las pápulas similares a queloides de AQN
INFLAMACIÓN REFRACTARIA
Algunos pacientes con AQN experimentan una progresión continuada de la enfermedad a pesar del tratamiento con las terapias anteriores. Se ha informado sobre el tratamiento exitoso de tales pacientes con cirugía, depilación láser o isotretinoína oral ; Sin embargo, los datos de eficacia son limitados.
Algunos pacientes con AQN experimentan una progresión continuada de la enfermedad a pesar del tratamiento con las terapias anteriores. Se ha informado sobre el tratamiento exitoso de tales pacientes con cirugía, depilación láser o isotretinoína oral ; Sin embargo, los datos de eficacia son limitados.
CIRUGÍA
Debido a que AQN generalmente se localiza, la extirpación quirúrgica puede usarse para extirpar el tejido afectado. La recurrencia es posible, particularmente si la escisión no es lo suficientemente profunda para eliminar los folículos capilares. La cirugía para AKN se analiza a continuación.
Debido a que AQN generalmente se localiza, la extirpación quirúrgica puede usarse para extirpar el tejido afectado. La recurrencia es posible, particularmente si la escisión no es lo suficientemente profunda para eliminar los folículos capilares. La cirugía para AKN se analiza a continuación.
DEPILACIÓN CON LÁSER
Un estudio piloto en el que se les dio 16 pacientes con AQN cinco tratamientos con láser con una larga pulsado de neodimio 1064 nm: granate de itrio y aluminio (Nd: YAG) láser a intervalos de cuatro semanas (fluencia de 35 a 45 J / cm 2 , duración del pulso de 10 a 30 mseg) sugiere el beneficio de la depilación láser para AKN [ 3] Se detectó mejoría en todos los pacientes, y el estudio encontró una disminución estadísticamente significativa en el recuento de pápulas, el recuento de placa y el tamaño de placa después del tratamiento. También se observó un ablandamiento significativo de las placas queloides. Los mejores resultados se observaron en pacientes con lesiones papulares tempranas en comparación con la enfermedad en estadio tardío con placas queloides, lo que sugiere que dicho tratamiento con láser es más beneficioso antes del desarrollo de la enfermedad avanzada. La recurrencia de AKN activa (caracterizada por unas pocas lesiones papulares) se desarrolló en 2 de 10 pacientes que fueron reevaluados después de un año; sin embargo, estos pacientes respondieron bien al tratamiento láser adicional.
Un estudio piloto en el que se les dio 16 pacientes con AQN cinco tratamientos con láser con una larga pulsado de neodimio 1064 nm: granate de itrio y aluminio (Nd: YAG) láser a intervalos de cuatro semanas (fluencia de 35 a 45 J / cm 2 , duración del pulso de 10 a 30 mseg) sugiere el beneficio de la depilación láser para AKN [ 3] Se detectó mejoría en todos los pacientes, y el estudio encontró una disminución estadísticamente significativa en el recuento de pápulas, el recuento de placa y el tamaño de placa después del tratamiento. También se observó un ablandamiento significativo de las placas queloides. Los mejores resultados se observaron en pacientes con lesiones papulares tempranas en comparación con la enfermedad en estadio tardío con placas queloides, lo que sugiere que dicho tratamiento con láser es más beneficioso antes del desarrollo de la enfermedad avanzada. La recurrencia de AKN activa (caracterizada por unas pocas lesiones papulares) se desarrolló en 2 de 10 pacientes que fueron reevaluados después de un año; sin embargo, estos pacientes respondieron bien al tratamiento láser adicional.
La mejora después del tratamiento con un láser de Nd: YAG de 1064 nm de pulso largo también se ha documentado en un informe de caso [ 45 ]. El tratamiento con otros láseres de depilación también puede ser efectivo; al menos dos pacientes han mejorado después del tratamiento con un láser de diodo de 810 nm [ 46 ].
La reducción de la densidad del cabello en el sitio de tratamiento es un efecto secundario esperado de este tratamiento. Otros posibles efectos secundarios de la terapia con láser incluyen la dispigmentación, formación de ampollas y cicatrices.
ISOTRETINOÍNA ORAL
Aunque los informes de casos sugieren que la isotretinoína oral puede ayudar a algunos pacientes con inflamación severa resistente al tratamiento, los datos limitados sobre la eficacia y los múltiples efectos secundarios de la isotretinoína (teratogenicidad, hiperlipidemia, xerosis, cambios visuales, etc.) evitan una recomendación para uso de rutina para AQN. Un informe de un caso describe a un hombre con queratosis folicular de spinvacaldecalvans y AQN que experimentó mejoría en AQN durante el tratamiento con 20 mg de isotretinoína oral al día (0,25 mg / kg por día) [ 34].] El AQN en el vértice demostró una mejoría rápida de la inflamación en cuestión de semanas; sin embargo, la reducción de la inflamación en el área nucal fue menos dramática. La mejoría se mantuvo durante al menos un año con una dosis reducida de isotretinoína (20 mg cada dos o tres días).
Aunque los informes de casos sugieren que la isotretinoína oral puede ayudar a algunos pacientes con inflamación severa resistente al tratamiento, los datos limitados sobre la eficacia y los múltiples efectos secundarios de la isotretinoína (teratogenicidad, hiperlipidemia, xerosis, cambios visuales, etc.) evitan una recomendación para uso de rutina para AQN. Un informe de un caso describe a un hombre con queratosis folicular de spinvacaldecalvans y AQN que experimentó mejoría en AQN durante el tratamiento con 20 mg de isotretinoína oral al día (0,25 mg / kg por día) [ 34].] El AQN en el vértice demostró una mejoría rápida de la inflamación en cuestión de semanas; sin embargo, la reducción de la inflamación en el área nucal fue menos dramática. La mejoría se mantuvo durante al menos un año con una dosis reducida de isotretinoína (20 mg cada dos o tres días).
En un segundo informe de caso, seis meses de isotretinoína oral (dosis inicial de 0,6 mg / kg por día durante tres meses, luego 0,3 mg / kg por día durante tres meses) se asociaron con la resolución de pápulas en un paciente con AKN [ 47 ]. Una recurrencia después del final del tratamiento respondió a un segundo ciclo de isotretinoína (0.1 mg / kg por día).
La isotretinoína es teratogénica y no debe administrarse a mujeres embarazadas. En los Estados Unidos, se requiere la participación en el programa iPLEDGE , un programa de administración de riesgos basado en computadora diseñado para reducir la exposición fetal a la isotretinoína, para la prescripción del medicamento. Los múltiples efectos adversos de la isotretinoína se revisan por separado.
TRATAMIENTO DE LA CICATRIZACIÓN QUELOIDEA
Las principales opciones terapéuticas para el tratamiento de cicatrices queloideas en AKN son las inyecciones intralesionales de corticosteroides y la cirugía.
Las principales opciones terapéuticas para el tratamiento de cicatrices queloideas en AKN son las inyecciones intralesionales de corticosteroides y la cirugía.
PÁPULAS Y PLACAS PEQUEÑAS
Nuestro tratamiento de primera línea preferido para las cicatrices queloides pequeñas es la inyección intralesional de corticosteroides.
Nuestro tratamiento de primera línea preferido para las cicatrices queloides pequeñas es la inyección intralesional de corticosteroides.
CORTICOSTEROIDES INTRALESIONALES
Al igual que con los queloides verdaderos, la inyección intralesional de corticosteroides puede ser útil para suavizar y contraer pápulas queloides o placas de AQN.
Al igual que con los queloides verdaderos, la inyección intralesional de corticosteroides puede ser útil para suavizar y contraer pápulas queloides o placas de AQN.
Normalmente utilizamos acetónido de triamcinolona en concentraciones de 5 a 40 mg / ml, dependiendo del tamaño del área con cicatrices. Las pápulas queloides pueden mejorar con dosis tan bajas como 5 mg / ml, mientras que las placas grandes y gruesas pueden requerir dosis más altas para lograr una mejora notable. Las inyecciones pueden repetirse cada cuatro a seis semanas y la dosis puede graduarse progresivamente en función de la respuesta. Se espera al menos una respuesta clínica parcial (al menos cierto ablandamiento de las lesiones) dentro de uno a tres tratamientos. (Ver "Inyección intralesional" ).
Las inyecciones intralesionales pueden ser incómodas. Cuando sea necesario, la aplicación de un analgésico tópico (p. Ej., Lidocaína tópica ) antes del tratamiento puede ayudar a reducir la incomodidad. Alternativamente, el corticosteroide puede diluirse en lidocaína. Pulverizar el sitio con nitrógeno líquido antes de la inyección también puede ayudar a reducir el dolor de la inyección y puede facilitar la inyección en el tejido; sin embargo, la hipopigmentación es un riesgo de nitrógeno líquido, particularmente en individuos con piel tipo IV a VI. Por lo general, no utilizamos nitrógeno líquido para la analgesia.
PLACAS MÁS GRANDES
Los pacientes con cicatrices extensas o cicatrices que no mejoran adecuadamente con el tratamiento con corticosteroides intralesionales pueden beneficiarse de la terapia quirúrgica para extirpar el área afectada.
Los pacientes con cicatrices extensas o cicatrices que no mejoran adecuadamente con el tratamiento con corticosteroides intralesionales pueden beneficiarse de la terapia quirúrgica para extirpar el área afectada.
CIRUGÍA
Las terapias quirúrgicas están indicadas para el tratamiento de cicatrices extensas que se observan en la placa o la etapa tumoral AKN. Una vez que se han desarrollado placas grandes, las opciones quirúrgicas son el único recurso efectivo. La cirugía también se puede utilizar para eliminar el sitio de la enfermedad en pacientes con enfermedad activa que responde poco al tratamiento médico. (Ver "inflamación refractaria" más arriba).
Las terapias quirúrgicas están indicadas para el tratamiento de cicatrices extensas que se observan en la placa o la etapa tumoral AKN. Una vez que se han desarrollado placas grandes, las opciones quirúrgicas son el único recurso efectivo. La cirugía también se puede utilizar para eliminar el sitio de la enfermedad en pacientes con enfermedad activa que responde poco al tratamiento médico. (Ver "inflamación refractaria" más arriba).
● Procedimientos : AQN se ha tratado con éxito con la escisión del escalpelo, la escisión electroquirúrgica y la escisión con láser. Independientemente de la técnica quirúrgica seleccionada, las siguientes medidas pueden optimizar los resultados:
• Escisión profunda : la profundidad de la escisión debe extenderse por debajo de la base de los folículos capilares para reducir el riesgo de recurrencia [ 11,48,49 ]. Algunos autores sugieren extirpación al nivel del tejido subcutáneo profundo o de la fascia muscular, dada la propensión de los folículos capilares a extenderse a la grasa subcutánea [ 48 ].
• Escisión horizontal : orientar la escisión horizontalmente con la intención de incorporar la línea de implantación posterior puede producir los mejores resultados estéticos [ 48 ].
Las opciones para el cierre de las heridas por escisión incluyen el cierre primario y la curación por segunda intención.
• Cierre primario de la herida: el cierre primario de la herida proporciona un cierre inmediato de la herida y simplifica el cuidado de la herida para el paciente. Sin embargo, la alta tensión en el cierre puede provocar la diseminación de la cicatriz, cicatrización hipertrófica o movimiento restringido de la cabeza. Por lo tanto, la evaluación preoperatoria debe incluir una evaluación del tamaño de la escisión y la laxitud de la piel circundante para determinar si la herida se puede cerrar cómodamente. Si el área afectada es demasiado grande para extirpar en un solo procedimiento, la escisión por etapas con cierre primario o curación por intención secundaria son opciones [ 48 ].
• Curación por intención secundaria : la curación por intención secundaria elimina la preocupación por la tensión excesiva de la herida que puede ocurrir con el cierre primario de la herida, pero requiere un promedio de 6 a 12 semanas para que la herida se cierre. Las cicatrices a menudo se contraen a un tamaño más pequeño que el área escindida inicial, produciendo cosmesis mejorada después de la cicatrización [ 50 ]. Una revisión encontró datos que sugieren que ocurren menos recurrencias con la curación por intención secundaria que con el cierre primario; sin embargo, sería necesario realizar más estudios para confirmar este hallazgo [ 50 ]. Los autores postularon que el proceso de curación de la intención secundaria podría eliminar efectivamente el tejido peludo o que la respuesta inflamatoria o el proceso de curación prolongada podrían tener otros efectos beneficiosos sobre AQN [ 50].
También se ha descrito el injerto de piel de grosor dividido, pero se considera menos que óptimo, ya que la cicatriz suele ser deprimida y texturalmente diferente (brillante y lisa) en comparación con la piel circundante [ 48 ]. Además, el color de la piel injertada generalmente no coincide con la piel circundante.
● Evidencia : aunque la cirugía se considera un tratamiento efectivo para AKN, pocos estudios han evaluado su eficacia. Uno de los estudios más grandes que evaluaron la intervención quirúrgica para AKN involucró a 25 hombres con AKN refractarios a la terapia médica [ 48].] Los pacientes tenían extensas placas queloides coalescentes en el cuero cabelludo nucal. Veinte pacientes se sometieron a una escisión completa con cierre primario del defecto quirúrgico. El resto tenía lesiones demasiado grandes para este procedimiento y tenía una escisión en dos etapas con cierre primario (cuatro pacientes) o excisión con cicatrización de intención secundaria (un paciente). Un año después del procedimiento, tanto el cirujano como los pacientes evaluaron los resultados cosméticos como buenos o excelentes. Las complicaciones incluyeron recidiva leve de pápulas y pústulas pequeñas en 15 pacientes (manejadas con corticosteroides tópicos) y cicatrices hipertróficas en cuatro pacientes que tuvieron extirpación de lesiones grandes y cierre primario de la herida en un solo procedimiento. En este estudio particular, los cuatro pacientes que se sometieron a la escisión electroquirúrgica parecían tener un requerimiento mayor de medicación para el dolor que los pacientes cuyas escisiones se realizaron con un bisturí. Sin embargo, también se ha informado una buena tolerancia a la electrocirugía [8].
El tratamiento exitoso con escisión láser de dióxido de carbono seguido de curación de intención secundaria se describió en una serie que incluyó a seis pacientes tratados con esta modalidad [ 49 ]. Ningún paciente experimentó recurrencias durante los períodos de seguimiento de 3 a 47 meses. Dos pacientes desarrollaron cicatrización hipertrófica leve que respondió bien a la inyección intralesional de corticosteroides.
● INTERVENCIONES POSQUIRÚRGICAS
Las inyecciones intralesionales de corticosteroides son útiles para ablandar y aplacar las cicatrices hipertróficas y se pueden utilizar si se desarrollan cicatrices después de la cirugía [ 51 ]. Algunos autores sugieren el uso de inyecciones de corticosteroides intralesionales comenzando en el momento de la extracción de la sutura de las heridas suturadas, como suele hacerse después de la escisión de queloides verdaderos [ 36 ]. Sin embargo, el efecto de las inyecciones profilácticas de corticosteroides intralesionales en los resultados de los pacientes no se ha evaluado específicamente para AKN. Es de destacar que las inyecciones intralesionales de corticosteroides pueden inhibir la contracción de la herida y no deben realizarse en heridas que cicatrizarán por segunda intención. (Consulte "Queloides y cicatrices hipertróficas", sección sobre "Excisión" ).
Las inyecciones intralesionales de corticosteroides son útiles para ablandar y aplacar las cicatrices hipertróficas y se pueden utilizar si se desarrollan cicatrices después de la cirugía [ 51 ]. Algunos autores sugieren el uso de inyecciones de corticosteroides intralesionales comenzando en el momento de la extracción de la sutura de las heridas suturadas, como suele hacerse después de la escisión de queloides verdaderos [ 36 ]. Sin embargo, el efecto de las inyecciones profilácticas de corticosteroides intralesionales en los resultados de los pacientes no se ha evaluado específicamente para AKN. Es de destacar que las inyecciones intralesionales de corticosteroides pueden inhibir la contracción de la herida y no deben realizarse en heridas que cicatrizarán por segunda intención. (Consulte "Queloides y cicatrices hipertróficas", sección sobre "Excisión" ).
Los estudios han arrojado resultados mixtos sobre la eficacia del imiquimod tópico para la prevención de la recurrencia posquirúrgica de queloides verdaderos [ 51 ]. El valor de esta intervención en AKN no está claro.
TERAPIA EMERGENTE
LA LUZ ULTRAVIOLETA
La luz ultravioleta (UV) puede inducir efectos anti-fibróticos en la piel mediante la inducción de la producción de metaloproteinasa de la matriz, así como efectos antiinflamatorios [ 52 ]. Los hallazgos de un pequeño ensayo aleatorizado dividido en el cuero cabelludo de pacientes con lesiones papulares de AQN sugieren que la fototerapia UVB dirigida puede ser una terapia útil [ 52] Los pacientes fueron asignados aleatoriamente al tratamiento con UVB dirigido tres veces por semana al área afectada en el lado derecho o izquierdo del cuero cabelludo durante ocho semanas. El dispositivo UVB emitió una luz de 290 nm a 320 nm, con picos a 303 nm y 313 nm. Después de ocho semanas, se trataron ambos lados del cuero cabelludo. Después del período inicial de ocho semanas, el recuento medio de lesiones en los lados tratados disminuyó significativamente, mientras que los recuentos en el lado no tratado no cambiaron. Se demostró una mejoría adicional en los sitios que recibieron ocho semanas adicionales de tratamiento con UVB.
La luz ultravioleta (UV) puede inducir efectos anti-fibróticos en la piel mediante la inducción de la producción de metaloproteinasa de la matriz, así como efectos antiinflamatorios [ 52 ]. Los hallazgos de un pequeño ensayo aleatorizado dividido en el cuero cabelludo de pacientes con lesiones papulares de AQN sugieren que la fototerapia UVB dirigida puede ser una terapia útil [ 52] Los pacientes fueron asignados aleatoriamente al tratamiento con UVB dirigido tres veces por semana al área afectada en el lado derecho o izquierdo del cuero cabelludo durante ocho semanas. El dispositivo UVB emitió una luz de 290 nm a 320 nm, con picos a 303 nm y 313 nm. Después de ocho semanas, se trataron ambos lados del cuero cabelludo. Después del período inicial de ocho semanas, el recuento medio de lesiones en los lados tratados disminuyó significativamente, mientras que los recuentos en el lado no tratado no cambiaron. Se demostró una mejoría adicional en los sitios que recibieron ocho semanas adicionales de tratamiento con UVB.
La terapia con UVB fue bien tolerada; los efectos secundarios fueron leves, incluyendo sensación de ardor transitorio, eritema e hiperpigmentación. Sin embargo, la necesidad de visitas clínicas múltiples puede hacer que la fototerapia sea inconveniente para algunos pacientes.
RADIOTERAPIA
La radioterapia condujo a la resolución de la enfermedad activa en un paciente con AKN refractario al tratamiento [ 53 ]. La enfermedad no se repitió durante un período de seguimiento de 20 meses.
La radioterapia condujo a la resolución de la enfermedad activa en un paciente con AKN refractario al tratamiento [ 53 ]. La enfermedad no se repitió durante un período de seguimiento de 20 meses.
Fuente UpToDate
New England Journal of Medicine
REFERENCIAS
1) Kaposi M. Über die sogenannte Framboesia und mehrere andere Arten von papillaren neubildungel der Haut. Arch Dermatol Syphilol 1869; 1:382.
2) Adamson HG. Dermatitis papillaris capillittii (Kaposi). Acne keloid. Br J Dermatol 1914; 26:69.
3) Esmat SM, Abdel Hay RM, Abu Zeid OM, Hosni HN. The efficacy of laser-assisted hair removal in the treatment of acne keloidalis nuchae; a pilot study. Eur J Dermatol 2012; 22:645.
4) Quarles FN, Brody H, Badreshia S, et al. Acne keloidalis nuchae. Dermatol Ther 2007; 20:128.
5) Alexis A, Heath CR, Halder RM. Folliculitis keloidalis nuchae and pseudofolliculitis barbae: are prevention and effective treatment within reach? Dermatol Clin 2014; 32:183.
6) Ross EK, Shapiro J. Primary Cicatricial Alopecia. In: Hair growth and disorders, Blume-Peytavi U, Whiting DA, Trüb RM (Eds), Springer, Berlin 2008. p.187.
7) McMichael A, Guzman Sanchez D, Kelly P. Folliculitis & the Follicular Occlusion Tetrad. In: Dermatology, 2nd ed, Bolognia JL, Jorizzo JL, Rapini RP, et al. (Eds), Elsevier Mosby, St. Louis 2008. p.517.
8) Beckett N, Lawson C, Cohen G. Electrosurgical excision of acne keloidalis nuchae with secondary intention healing. J Clin Aesthet Dermatol 2011; 4:36.
9) Knable AL Jr, Hanke CW, Gonin R. Prevalence of acne keloidalis nuchae in football players. J Am Acad Dermatol 1997; 37:570.
10) Salami T, Omeife H, Samuel S. Prevalence of acne keloidalis nuchae in Nigerians. Int J Dermatol 2007; 46:482.
11) Glenn MJ, Bennett RG, Kelly AP. Acne keloidalis nuchae: treatment with excision and second-intention healing. J Am Acad Dermatol 1995; 33:243.
12) Dunwell P, Rose A. Study of the skin disease spectrum occurring in an Afro-Caribbean population. Int J Dermatol 2003; 42:287.
13) George AO, Akanji AO, Nduka EU, et al. Clinical, biochemical and morphologic features of acne keloidalis in a black population. Int J Dermatol 1993; 32:714.
14) Adegbidi H, Atadokpede F, do Ango-Padonou F, Yedomon H. Keloid acne of the neck: epidemiological studies over 10 years. Int J Dermatol 2005; 44 Suppl 1:49.
15) Ogunbiyi A, George A. Acne keloidalis in females: case report and review of literature. J Natl Med Assoc 2005; 97:736.
16) Dinehart SM, Tanner L, Mallory SB, Herzberg AJ. Acne keloidalis in women. Cutis 1989; 44:250.
17) Kelly AP. Pseudofolliculitis barbae and acne keloidalis nuchae. Dermatol Clin 2003; 21:645.
18) Khumalo NP, Jessop S, Gumedze F, Ehrlich R. Hairdressing and the prevalence of scalp disease in African adults. Br J Dermatol 2007; 157:981.
19) Kundu RV, Patterson S. Dermatologic conditions in skin of color: part II. Disorders occurring predominately in skin of color. Am Fam Physician 2013; 87:859.
20) Dinehart SM, Herzberg AJ, Kerns BJ, Pollack SV. Acne keloidalis: a review. J Dermatol Surg Oncol 1989; 15:642.
21) Smith JD, Odom RB. Pseudofolliculitis capitis. Arch Dermatol 1977; 113:328.
22) Kenney JA Jr. Dermatoses: common in blacks. Postgrad Med 1977; 61:122.
23) Halder RM. Hair and scalp disorders in blacks. Cutis 1983; 32:378.
24) Basler RS. Acne mechanica in athletes. Cutis 1992; 50:125.
25) Cosman B, Wolff M. Acne keloidalis. Plast Reconstr Surg 1972; 50:25.
26) Herzberg AJ, Dinehart SM, Kerns BJ, Pollack SV. Acne keloidalis. Transverse microscopy, immunohistochemistry, and electron microscopy. Am J Dermatopathol 1990; 12:109.
27) Sperling LC, Homoky C, Pratt L, Sau P. Acne keloidalis is a form of primary scarring alopecia. Arch Dermatol 2000; 136:479.
28) Ferguson RA, Yu H, Kalyvas M, et al. Ultrasensitive detection of prostate-specific antigen by a time-resolved immunofluorometric assay and the Immulite immunochemiluminescent third-generation assay: potential applications in prostate and breast cancers. Clin Chem 1996; 42:675.
29) Azurdia RM, Graham RM, Weismann K, et al. Acne keloidalis in caucasian patients on cyclosporin following organ transplantation. Br J Dermatol 2000; 143:465.
30) Carnero L, Silvestre JF, Guijarro J, et al. Nuchal acne keloidalis associated with cyclosporin. Br J Dermatol 2001; 144:429.
31) Piaserico S, Fortina AB, Cavallini F, Alaibac M. Acne keloidalis of the scalp in a renal transplant patient treated with cyclosporine. Acta Derm Venereol 2009; 89:312.
32) Pirmez R, Price VH, Cuzzi T, Trope BM. Acne keloidalis nuchae in renal transplant patients receiving tacrolimus and sirolimus. Australas J Dermatol 2016; 57:156.
33) Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol 2005; 53:1.
34) Goh MS, Magee J, Chong AH. Keratosis follicularis spinulosa decalvans and acne keloidalis nuchae. Australas J Dermatol 2005; 46:257.
35) Janjua SA, Iftikhar N, Pastar Z, Hosler GA. Keratosis follicularis spinulosa decalvans associated with acne keloidalis nuchae and tufted hair folliculitis. Am J Clin Dermatol 2008; 9:137.
36) McMichael A, Curtis AR, Guzman-Sanchez D, Kelly AP. Folliculitis and other follicular disorders. In: Dermatology, 3rd ed, Bolognia JL, Jorizzo JL, Schaffer JV (Eds), Elsevier, Philadelphia; London 2012. Vol 1, p.571.
37) Vasily DB, Breen PC, Miller OF 3rd. Acne keloidalis nuchae: report and treatment of a severe case. J Dermatol Surg Oncol 1979; 5:228.
38) Hodak E, Feinmesser M, Segal T, et al. Follicular cutaneous T-cell lymphoma: a clinicopathological study of nine cases. Br J Dermatol 1999; 141:315.
39) Battistella M, Beylot-Barry M, Bachelez H, et al. Primary cutaneous follicular helper T-cell lymphoma: a new subtype of cutaneous T-cell lymphoma reported in a series of 5 cases. Arch Dermatol 2012; 148:832.
40) Maranda EL, Simmons BJ, Nguyen AH, et al. Treatment of Acne Keloidalis Nuchae: A Systematic Review of the Literature. Dermatol Ther (Heidelb) 2016; 6:363.
41) Shapero J, Shapero H. Acne keloidalis nuchae is scar and keloid formation secondary to mechanically induced folliculitis. J Cutan Med Surg 2011; 15:238.
42) Callender VD, Young CM, Haverstock CL, et al. An open label study of clobetasol propionate 0.05% and betamethasone valerate 0.12% foams in the treatment of mild to moderate acne keloidalis. Cutis 2005; 75:317.
43) Gregoriou S, Kritsotaki E, Katoulis A, Rigopoulos D. Use of tazarotene foam for the treatment of acne vulgaris. Clin Cosmet Investig Dermatol 2014; 7:165.
44) Eichenfield LF, Krakowski AC, Piggott C, et al. Evidence-based recommendations for the diagnosis and treatment of pediatric acne. Pediatrics 2013; 131 Suppl 3:S163.
45) Dragoni F, Bassi A, Cannarozzo G, et al. Successful treatment of acne keloidalis nuchae resistant to conventional therapy with 1064-nm ND:YAG laser. G Ital Dermatol Venereol 2013; 148:231.
46) Shah GK. Efficacy of diode laser for treating acne keloidalis nuchae. Indian J Dermatol Venereol Leprol 2005; 71:31.
47) Stieler W, Senff H, Jänner M. [Folliculitis nuchae scleroticans--successful treatment with 13-cis-retinoic acid (isotretinoin)]. Hautarzt 1988; 39:739.
48) Gloster HM Jr. The surgical management of extensive cases of acne keloidalis nuchae. Arch Dermatol 2000; 136:1376.
49) Kantor GR, Ratz JL, Wheeland RG. Treatment of acne keloidalis nuchae with carbon dioxide laser. J Am Acad Dermatol 1986; 14:263.
50) Bajaj V, Langtry JA. Surgical excision of acne keloidalis nuchae with secondary intention healing. Clin Exp Dermatol 2008; 33:53.
51) Monstrey S, Middelkoop E, Vranckx JJ, et al. Updated scar management practical guidelines: non-invasive and invasive measures. J Plast Reconstr Aesthet Surg 2014; 67:1017.
52) Okoye GA, Rainer BM, Leung SG, et al. Improving acne keloidalis nuchae with targeted ultraviolet B treatment: a prospective, randomized, split-scalp comparison study. Br J Dermatol 2014; 171:1156.
53) Millán-Cayetano JF, Repiso-Jiménez JB, Del Boz J, de Troya-Martín M. Refractory acne keloidalis nuchae treated with radiotherapy. Australas J Dermatol 2017; 58:e11.
Estudió Medicina en Universidad Estatal de Cuenca
ACNÉ QUELOIDEO / FOLICULITIS ESCLEROSANTE DE LA NUCA (ACNE KELOIDALIS NUCHAE)
INTRODUCCIÓN - El acné queloide nucal (AQN) es un trastorno crónico común que involucra la