![]() |
| Hospital "Dr. Ángel Pintos" de Azul. |
Paciente de 40 años con diagnóstico de infección por HIV desde 2002. En el año 2004 comenzó tratamiento antirretroviral (LPN/RTV/AZT/3TC) por disminución del nivel de CD4 (menos de 200/mm3) pero sin haber presentado enfermedad marcadora.
Lesiones de sarcoma de Kaposi sobre un fondo de linfedema severo.
Las mismas lesiones de la imagen superior en una vista ampliada
Lesiones de Kaposi con linfedema. Se observan lesiones hiperqueratósicas interdigitales asociadas a Kaposi crónico y sobreinfección crónica por hongos y bacterias.
Lesiones de Kaposi plantares con evolución a hiperqueratosis y agrietamiento profundo que dificulta la deambulación. Tendencia a las formas vegetantes.
Se observa la tendencia a lesiones proliferativas vegetantes.
Las sobreinfecciones fúngicas crónicas favorecidas por el severo inmunocompromiso se combinan con las lesiones proliferativas del SK.
Linfedema y signo de Kaposi-Stemmer muestra la imposibilidad de producir un pliegue en la piel entre los dedos.
Signo de Kaposi-Stemmer
Lesiones de Kaposi en escroto.
Desde entonces ha tenido poca adherencia al tratamiento que hace aproximadamente un año tiene suspendido. Desde hace algunos años presentó lesiones en piel de tipo maculo papular en brazos que biopsiadas fueron informadas como sarcoma de Kaposi. Las mismas se mantuvieron estables y posteriormente con tendencia involutiva mientras realizaba tratamiento antirretroviral. Desde hace 1 año estas lesiones se mantuvieron en miembros superiores pero aparecieron nuevas en ambos miembros inferiores desde la raíz de los mismos. Las lesiones tienen aspecto papular, son relativamente duras de color violáceas a marrón oscuras , de forma redondeada a ovales y desde algunos mm hasta algunos centímetros de diámetro. Desde hace 6 meses las lesiones adoptaron el aspecto hiperqueratósicas en los pies con tendencia a la adopción de formas vegetantes, despidiendo olor fétido sobre todo en las plantas de los pies. Se agregó en este tiempo la presencia de severo linfedema bilateral que dificulta la marcha y que altera severamente la calidad de vida.
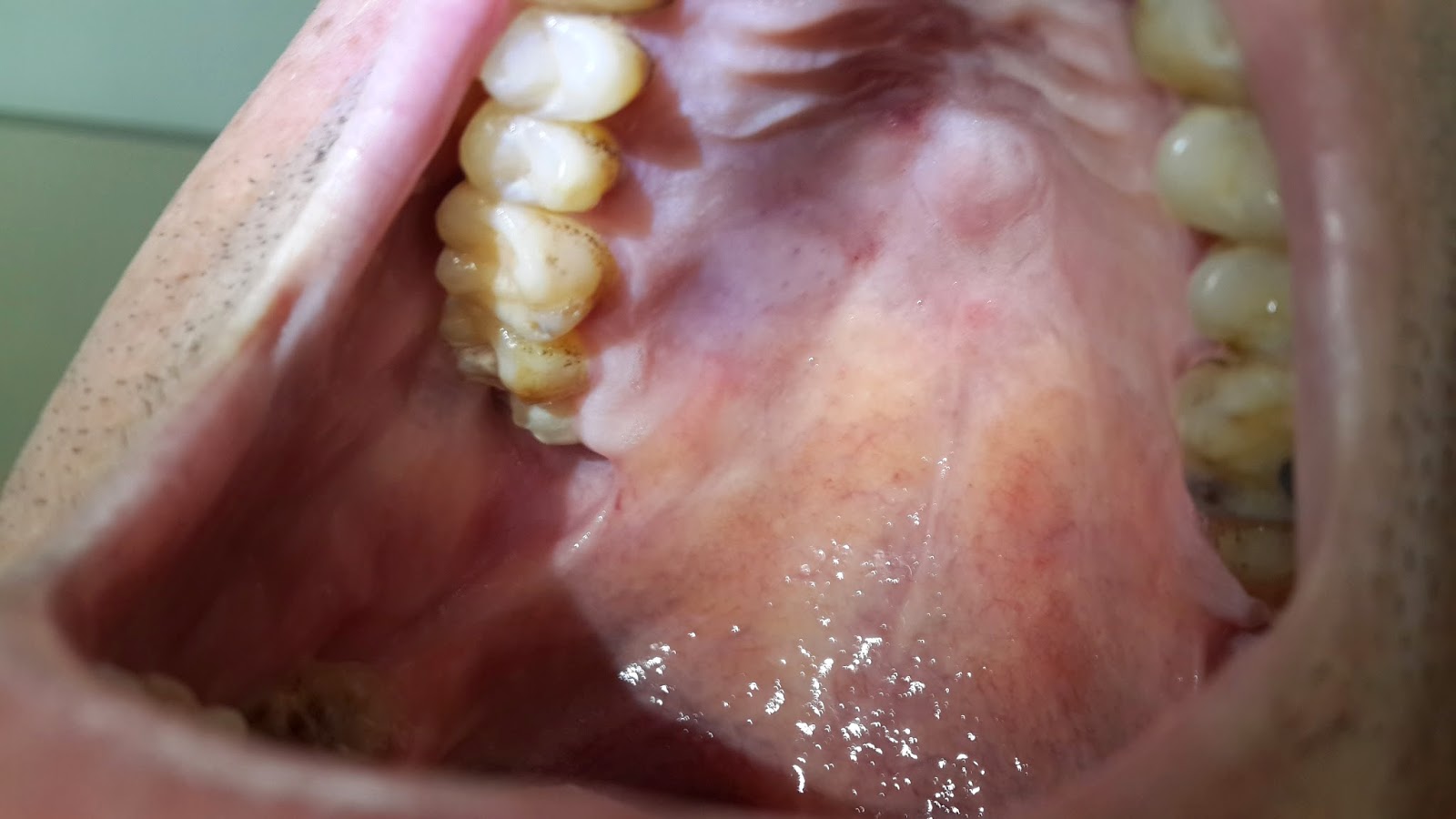
El paladar duro se observa una lesión mucosa de aspecto Kaposiforme.
Lesiones de aspecto Kaposiforme en paladar duro.
Otra imagen de paladar duro.
En miembro superior derecho presenta una sola lesión que es la originalmente biopsiada.
Se palpan algunas adenopatías en región laterocervical de alrededor de 1 cm omenos de diámetro, móviles.
No refiere síndrome de impregnación general, y sólo dice haber tenido sensación febril aunque no se cuantificó la temperatura hace 72 horas coincidiendo con el comienzo de una tos escasamente productiva.
Último valor de CD4 que figura en la historia clínica es de julio de 2011 y es de 29 células/mm3 con carga viral no detectable en ese momento en el que recibía tratamiento ARV(6 meses antes la carga viral había sido de 108.404 copias /ml y el dosaje de CD4 de 2 células/ml.
Se solicitó carga viral,CD4, análisis completos de sangre y Rx de tórax F y P.
SARCOMA DE KAPOSI ASOCIADO AL SIDA
INTRODUCCIÓN
El sarcoma de Kaposi (SK) es un tumor vascular que está etiológicamente asociado con el herpesvirus humano 8 (HHV-8), que también se conoce como el herpesvirus asociado-SK (VHSK) [1].
EPIDEMIOLOGÍA Y FACTORES DE RIESGO
Hay cuatro formas epidemiológicas de SK, todos los cuales han sido relacionados con la infección por HHV-8. Estos incluyen:
- SARCOMA DE KAPOSI RELACIONADO CON EL SIDA o SK epidémico es el tumor más común que surge en las personas infectadas por el VIH. El SK se considera una enfermedad marcadora de SIDA (CDC) pero su incidencia ha disminuido sustancialmente desde la era de la terapia de alta intensidad (TARGA).
- SARCOMA DE KAPOSI ENDÉMICO O AFRICANO. El SK era endémico en toda África ecuatorial, en particular en el África subsahariana, con anterioridad a la epidemia del VIH. Desde el comienzo de la epidemia del VIH, la incidencia de SK se ha incrementado dramáticamente en esas regiones [3,4].
- SARCOMA DE KAPOSI ASOCIADO CON EL TRASPLANTE DE ÓRGANOS. La incidencia de SK se incrementa después de un trasplante de órgano sólido, presumiblemente, al menos parcialmente, debido a la inmunosupresión crónica. Además, el propio trasplante puede transmitir HHV-8 infección. La presentación clínica en esta configuración es similar a la de SK clásicos.
- SARCOMA DE KAPOSI CLÁSICO. Es una enfermedad proliferativa cutánea indolente que afecta principalmente a los hombres mayores de Mediterráneo y de origen judío [5,6].
EPIDEMIOLOGÍA DE SK RELACIONADO CON EL SIDA
Aunque el SK ha sido reportado en todos los grupos de riesgo para la infección por el VIH, es más común en hombres homosexuales o bisexuales, y es mucho menos común en los heterosexuales o drogadictos por vía EV, o en receptores de transfusiones,mujeres y niños. [2].
En los pacientes con sarcoma de Kaposi relacionado con el SIDA, el recuento de CD4 parece ser el factor más importante asociado con el desarrollo de SK. En una serie de 70 pacientes que se presentaron con un nuevo diagnóstico de SK mientras estaban en tratamiento con terapia antirretroviral combinada (TAR), las tasas de desarrollo de SK para los pacientes con recuentos de CD4 menores a 200, de 200 a 349 y de 350 a 499 células / mm 3 fueron: 18.9, 3.6, y 4.1, respectivamente en comparación con aquellos con más de 500 células / mm3 [7].
En los países desarrollados, el SK relacionado con el SIDA es predominantemente una enfermedad de hombres. Por el contrario, en las zonas pobres en recursos, como África subsahariana, el SIDA relacionados con SK también es más frecuente en varones, aunque la diferencia es menos pronunciada.
IMPACTO DE LA TERAPIA ANTIRRETROVIRAL
Desde la introducción de la terapia antirretroviral combinada (TAR), la incidencia de SK ha disminuido notablemente en los pacientes infectados por el VIH [8-11]. Estudios epidemiológicos a gran escala han reportado una disminución similar en la incidencia de SK se correlaciona con la introducción de la terapia antirretroviral [9,12].
La incidencia de SK es especialmente alto en los primeros seis meses después del inicio de la terapia antirretroviral (TAR), y luego cae dramáticamente con la continuación del tratamiento [13]. La alta incidencia inmediatamente después del inicio del TAR puede ser atribuible a la inmunosupresión relativamente severa que llevó a la TAR o a la exacerbación del SK por el síndrome inflamatorio de reconstitución inmune.
La disminución de la incidencia de SK no puede atribuirse a una disminución de la incidencia de la infección por HHV-8. En un informe, la prevalencia de la infección por HHV-8 se mantuvo aproximadamente constante entre los hombres en San Francisco desde 1978 a 1979 (26,5 por ciento), de 1984 a 1985 (29,6 por ciento) y de 1995-1996 (26,4 por ciento) [14].
HERPES VIRUS HUMANO 8
Existe una fuerte relación entre el desarrollo de SK y la infección por HHV-8 y la infección por el VIH [15-17].
En un estudio llevado a cabo en San Francisco, la seropositividad para HHV-8 se observó en el 38 por ciento de los 593 hombres que tenían sexo con hombres (HSH) en comparación con ninguno de 195 hombres heterosexuales [15]. Entre los pacientes infectados por el VIH que se infectaron con el VIH y el HHV-8 al inicio del estudio, la probabilidad a 10 años de padecer SK fue de aproximadamente 50 por ciento. El riesgo a los 10 años fue de aproximadamente 30 por ciento en los hombres infectados por el VIH que no eran seropositivos para HHV-8 al inicio del estudio, mientras que no hubo casos de SK entre los hombres VIH-negativos. Este estudio se llevó a cabo antes del uso generalizado de la terapia antirretroviral de alto impacto (TARGA), y la frecuencia de la progresión de la infección por HHV-8 a SK parece haber disminuido desde entonces.
ESTEROIDES E INFECCIÓN.
El tratamiento con corticoides se ha asociado con la inducción de SK y la exacerbación de SK preexistentes en personas infectadas por el VIH, así como en pacientes no-SIDA que reciben corticosteroides por trasplante de órganos, enfermedades autoinmunes o enfermedades linfoproliferativas [18].
La asociación de corticosteroides con SK es importante debido a la frecuente utilización de estos agentes en pacientes infectados por el VIH con una variedad de trastornos como la trombocitopenia inmune (PTI) y la neumonía por Pneumocystis jirovecii. En estos pacientes, las lesiones de SK pueden retroceder tras la reducción o retirada de esteroides [18,19].
Las infecciones oportunistas también se han asociado con la inducción de KS y con la exacerbación de la preexistente KS similar a la descrita con la terapia con corticosteroides. Los altos niveles de citoquinas proinflamatorias, que se ha demostrado en el contexto de las infecciones oportunistas, pueden ser responsables de estos efectos sobre el SK
SARCOMA DE KAPOSI CUTÁNEO
Aunque el SK puede implicar prácticamente cualquier sitio en el cuerpo, la enfermedad cutánea es más común y es la presentación inicial habitual para SK.
Las manifestaciones clínicas - Las lesiones cutáneas del SK aparecen con mayor frecuencia en las extremidades inferiores, la cara (especialmente la nariz), la mucosa oral, y los genitales (figuras 1 a 11). Las lesiones son a menudo elípticas y pueden estar dispuestas en forma lineal a lo largo de las líneas de tensión de la piel; pueden estar distribuidas simétricamente. Las lesiones no son dolorosas ni pruriginosas y por lo general no producen necrosis de la piel suprayacente o estructuras subyacentes.
![]() |
Figura 1. Una placa eritematosa está presente en la nariz de este paciente con SK. |
![]() |
Figura 2 Múltiples placas violáceas en la cara. |
![]() |
| Figura 3 Placa violácea correspondiente a lesión por sarcoma de Kaposi. |
![]() |
Figura 4. Multiple placas ovales, violáceas en extremidades superiores y tronco en este paciente con Kaposi relacionado al SIDA. |
La variedad de colores asociados a estas lesiones se debe a su vascularización e incluye tonos de rosa, rojo, púrpura y marrón. Halos perilesionales amarillos ocasionalmente pueden ser vistos (imagen 2).
Las lesiones tempranas pueden ser fácilmente confundidas con púrpura, hematomas, angiomas, dermatofibromas o nevus. Más comúnmente, sin embargo, las lesiones de SK son papulares, que varían en tamaño desde varios milímetros a varios centímetros de diámetro. Con menos frecuencia, las lesiones de SK pueden ser de tipo placa, especialmente en las plantas de los pies y los muslos, o exofíticas y vegetantes con hiperqueratosis de de la piel que las recubren (figuras).
![]() |
Figura 5. Lesiones eritematosas en la pierna de este paciente con Kaposi. |
![]() |
Figura 6. A) Pápula eritematosa en lapierna de este paciente de 54 años, Afro-Americano. B) Vista más cercana del mismo paciente. Es más dificultoso identificar lesions de Kaposi en pacientes con piel oscura.
|
![]() |
Figura 7. Sarcoma de Kaposi relacionado al SIDA. Múltiples pápulas violáceas presentes en los miembros inferiores. |
![]() |
Figura 8. Pápula vascular consistente con Kaposi en pene. |
![]() |
Figura 9. Multiple placas eritematosas hiperqueratósicas presents en la planta delpie de este paciente con Kaposi. |
![]() |
Figura 10. Placa ulcerada presente en planta de pie. |
![]() |
Figura 11. Nódulos de sarcoma de Kaposi en la encía. |
![]() |
Figura 12. Nódulos de sarcoma de Kaposi en paladar. |
El linfedema, sobre todo en la cara, los genitales y extremidades inferiores puede estar fuera de proporción con el grado de la enfermedad y puede estar relacionados con obstrucción vascular por las linfadenopatía pero también por las citoquinas implicadas en la patogénesis del SK.
DIAGNÓSTICO DIFERENCIAL
Un diagnóstico presuntivo de SK cutáneo con frecuencia puede ser hecho por un observador entrenado. Sin embargo, las lesiones tempranas pueden ser fácilmente confundidas como se dijo antes con púrpura, hematomas, angiomas, dermatofibromas o nevus.
La angiomatosis bacilar es el diagnóstico diferencial más importante en el diagnóstico diferencial. La angiomatosis bacilar es causada por especies de Bartonella, una, fastidioso, bacilo gram-negativo de crecimiento lento, y se trata fácilmente con antibióticos. Las lesiones de la piel de la angiomatosis bacilar generalmente aparecen como numerosas pequeñas pápulas rojas a púrpura que pueden agrandarse gradualmente y evolucionar a grandes lesiones o nódulos pediculados que pueden ser friables. La erupción puede estar asociada con síntomas como fiebre, escalofríos, malestar general, dolor de cabeza y anorexia. La biopsia es especialmente importante para las lesiones atípicas que están asociadas a síntomas sistémicos o aparecen o progresan rápidamente, con el fin de descartar la angiomatosis bacilar.
SK y angiomatosis bacilar pueden ocurrir simultáneamente en el mismo paciente. Además, las lesiones de angiomatosis bacilar pueden estar asociadas a neovascularización y áreas sólidas de células fusiformes que imitan al SK. El diagnóstico de angiomatosis bacilar es establecido por la identificación de los organismos causales con tinción de plata de Warthin-Starry.
Con menos frecuencia el SK puede ser confundido con lesiones por Pneumocystis jirovecii extrapulmonares, incluso en ausencia infección pulmonar [20].
DIAGNÓSTICO
Aunque un diagnóstico presuntivo de SK a menudo se puede hacer basándose en la apariencia característica de las lesiones, esto debe ser confirmado por una biopsia siempre que sea posible. La biopsia es especialmente importante para las lesiones atípicas que están asociadas a síntomas sistémicos o aparecen o progresan rápidamente, con el fin de descartar angiomatosis bacilar.
PATOLOGÍA
Hay tres rasgos histológicos característicos de SK tanto en sitios cutáneos como viscerales: la angiogénesis, la inflamación y la proliferación. Las lesiones generalmente muestran dos grandes anomalías: espirales de células fusiformes con infiltración leucocitaria y neovascularización con proliferación aberrante de vasos pequeños.
Estos pequeños vasos carecen de una membrana basal y muestran un comportamiento con fugas y microhemorragias con depósitos de hemosiderina [21]. A medida que la enfermedad progresa, se desarrollan placas y a continuación evolucionan a la forma nodular. El patrón histológico característico de SK no difiere entre los 4 grupos epidemiológicos afectados.
ENFERMEDAD VISCERAL
El SK se ha observado en casi cualquier sitio visceral, incluidos los ganglios linfáticos, hígado, páncreas, corazón, testículos, médula ósea, hueso y músculo esquelético [22,23]. Los sitios más frecuentes de enfermedad no cutánea son la cavidad oral, el tracto gastrointestinal, y el sistema respiratorio. Sin embargo, la afectación visceral como manifestación inicial de SK es relativamente poco común. Además, la enfermedad visceral ahora parece ser mucho menos frecuente, dado el uso de la terapia antirretroviral [24].
CAVIDAD ORAL
La participación de la cavidad oral se presenta en aproximadamente un tercio de los pacientes con sarcoma de Kaposi y es el sitio inicial en un 15 por ciento. El odontólogo es a menudo el primero en identificar estas lesiones (Figuras 11 y 12). El diagnóstico de SK debe ser confirmado por biopsia siempre que sea posible.
![]() |
Figura 11. Nódulos de sarcoma de Kaposi en la encía. |
![]() |
| Figura 12. Nódulos de sarcoma de Kaposi en paladar. |
El sitio intraoral más comúnmente afectada es el paladar, seguido de la encía [25]. Lesiones intraorales pueden llegar fácilmente traumatizadas durante la masticación normal, lo cual puede resultar en dolor, sangrado, ulceración o infección secundaria. Las lesiones avanzadas pueden interferir con la nutrición y el habla.
La presencia o ausencia de síntomas de lesiones orales es a menudo un factor importante en las decisiones de tratamiento.
SISTEMA GASTROINTESTINAL
Antes de la introducción generalizada de la terapia antirretroviral, el tracto gastrointestinal participaba en aproximadamente el 40 por ciento de los pacientes con sarcoma de Kaposi en el diagnóstico inicial y en hasta el 80 por ciento en las autopsias.
La afectación gastrointestinal puede ocurrir en ausencia de enfermedad cutánea. Las lesiones gastrointestinales pueden ser asintomáticas o pueden causar pérdida de peso, dolor abdominal, náuseas y vómitos, hemorragia digestiva alta o más baja, mala absorción, obstrucción intestinal, y / o diarrea [26,27].
Lesiones gastrointestinales SK son fácilmente reconocidas por el endoscopista (Figura 13) [28].
![]() |
Figura 13. Lesiones gástricas de sarcoma de Kaposi en un paciente HIV vositivo que se presentó con hemorragia digestiva, lesiones endobronquiales y compromiso de piel, asociados a derrame pleural hemorrágico. |
Por lo general son nódulos hemorrágicos que pueden ser aislados o confluentes y pueden ocurrir en cualquier parte del tracto gastrointestinal (Figura 14) [22]. El diagnóstico de SK debe ser confirmado por biopsia siempre que sea posible, a pesar de las biopsias pueden no demostrar SK debido a que las lesiones tienden a ser submucosas. Lesiones de alto grado son más propensas a la invasión y diseminación.
![]() |
Figura 14. Sarcoma de Kaposi del tracto digestivo en un paciente con SIDA que se presentó como una gran masa anular circunferencial con obstrucción luminal de colon y recto.
|
SISTEMA RESPIRATORIO
La afectación pulmonar es frecuente en el SK relacionado con el SIDA. No nos detendremos a analizar las manifestaciones clínicas del SK del aparato respiratorio pero digamos que los pacientes afectados pueden presentar dificultad para respirar, fiebre, tos, hemoptisis o dolor en el pecho, o bien la afectación pulmonar puede ser un hallazgo asintomático observado por primera vez en una radiografía de tórax. Los hallazgos radiológicos son variables y pueden incluir imágenes nodulares, intersticiales y / o infiltrados alveolares, derrame pleural, hiliares y / o adenopatías mediastinales, o incluso un nódulo aislado.
Las lesiones del SK tienen un aspecto característico de las lesiones, rojo cereza ligeramente levantadas visto en broncoscopia, lo que puede dar lugar a un diagnóstico presuntivo de KS pulmonares. Aunque la broncoscopia y las imágenes radiológicas se correlacionan bastante bien, los pacientes que han SK documentados por broncoscopia en ocasiones tienen las radiografías de tórax normales.
ESTADIFICACIÓN
Los pacientes se clasifican en función de tres parámetros (tabla 1:
- Extensión de la enfermedad (T): Se considera T0 a la enfermedad limitada a la piel con mínima o ninguna participación de la cavidad oral. T1 se considera a pacientes que presentan linfedema asociado, más amplia participación de la cavidad oral u otra enfermedad visceral los cuales tienen un pronóstico pobre.
- Estado Inmunológico (I): Los pacientes con más de 200 CD4 se los considera I0, y a los que tienen menos de 200 CD4 se los considera I1 y tienen mal pronóstico.
- Severidad de la enfermedad sistémica (S): S1 se considera a los pacientes con infecciones oportunistas, aftas, síntomas B (fiebre, sudoración nocturna, pérdida significativa de peso, diarrea de más de dos semanas). Los que no tienen nada de esto se los considera S0.
EVALUACIÓN INICIAL
La evaluación inicial de un paciente con SK consiste en un examen físico completo con especial atención a aquellas zonas típicamente afectadas por la enfermedad, como las extremidades inferiores, la cara, la mucosa oral, los genitales, el tracto gastrointestinal y los pulmones.
La valoración de la afectación visceral es guiada por la sintomatología y las pruebas básicas de laboratorio:
1 Prueba de sangre oculta en heces es la forma más sencilla de detectar afectación gastrointestinal. La endoscopia suele reservarse para los pacientes que dan positivo para sangre oculta o tienen síntomas gastrointestinales.
2 Radiografía de tórax es útil para detectar lesiones pulmonares. La broncoscopía debe reservarse para aquellos con una radiografía anormal y síntomas respiratorios persistentes en ausencia de otra causa.
TAC de tórax de abdomen y pelvis no son normalmente necesarias.
El recuento de CD4 y la carga viral del VIH son importantes para la estadificación y el pronóstico, y por lo tanto puede ser útil en la toma de decisiones sobre el tratamiento.
TRATAMIENTO
Los principales objetivos del tratamiento son la paliación de los síntomas, la prevención de la progresión de la enfermedad, y la contracción del tumor para aliviar el edema, el compromiso de órganos, y el estrés psicológico [33].
Se recomienda el tratamiento sistémico con combinaciones potentes de antirretrovirales (TAR) para prácticamente todos los pacientes con sarcoma de Kaposi relacionado con el SIDA [33-35]. La necesidad de un tratamiento más allá del TAR y la elección entre las distintas opciones depende de la extensión de la enfermedad, la rapidez de crecimiento del tumor, la carga viral VIH-1 , el recuento de CD4, y la condición médica general del paciente [36].
La terapia dirigida localmente se utiliza a menudo para paliar los síntomas causados por un tumor específico o para tratar una desfiguración cosmética. La terapia sistémica se utiliza para la enfermedad más extensa, pero un mayor deterioro con quimioterapia asociada, a un sistema inmunológico que ya está gravemente comprometido debe evitarse siempre que sea posible.
Se recomienda TAR con combinación de antirretrovirales para prácticamente todos los pacientes con sarcoma de Kaposi relacionado con el SIDA, y puede ser el único tratamiento necesario en ausencia de indicaciones específicas para la quimioterapia asociada.
La introducción de la terapia antirretroviral se ha asociado con una disminución sustancial en la incidencia y severidad de SK recién diagnosticados en pacientes infectados por el VIH. Un estudio de base de datos francesa que incluyó 54.999 pacientes con más de 180.000 pacientes-año de seguimiento se encontró que la tasa de incidencia de nuevos casos de SK se redujo de 32 por cada 1.000 personas-año en el período 1993-1994 a 3 por 1000 personas-años después de 1999 [37 ]. Por otra parte, la incidencia de afectación visceral en la presentación se redujo de más del 50 por ciento a menos del 30 por ciento. Disminuciones dramáticas similares se han visto en otros estudios [38.39].
Los estudios observacionales indican que la historia natural de SK relacionado con el SIDA ha cambiado desde la introducción de la terapia antirretroviral, además de una disminución en la incidencia del SK [40,41]. Un estudio retrospectivo que analizó SK de una base de datos de 4.439 personas con infección por VIH, tanto antes TAR (1990 a 1996) y después de tar (1997 a 2002) encontró que la media de los niveles de CD4 y los niveles medios de ARN del VIH fueron similares en los 366 pacientes desde la era pre-TAR y en los 40 pacientes en la era post-TAR [41]. Sin embargo, el riesgo general de morir fue significativamente menor en la era post-TAR .
El síndrome de reconstitución inmune debido al control de la infección por VIH es la explicación más probable para este pronóstico alterado en lugar de un efecto directo sobre el tumor. Aunque los inhibidores de la proteasa del VIH tienen propiedades antiangiogénicas y bloquean el desarrollo y la progresión de las lesiones de SK [42], no había diferencia en la probabilidad de respuesta clínica asociada con el uso de estos agentes [40,43]. Además, la disminución de la incidencia de SK se ha observado con los regímenes de TAR que no contienen inhibidores de la proteasa [37].
Aunque el tratamiento con TAR provoca aumentos en los recuentos de CD4 por encima de los niveles típicamente asociados con una mayor susceptibilidad a la infección, algunos pacientes desarrollan SK relacionado con el VIH a pesar de la aparente corrección de su inmunodeficiencia [44].
SÍNDROME DE RECONSTITUCIÓN INMUNE(SIR).
El término "síndrome de reconstitución inmune " (SIR) es una serie de respuestas del huésped que pueden ocurrir después de la iniciación de la terapia antirretroviral. Además del empeoramiento de los síntomas de las infecciones preexistentes con SRI, la iniciación de la terapia antirretroviral se ha asociado con la progresión de SK dentro de tres a seis semanas después de comenzada la terapia antirretroviral [45,46].
Los factores asociados con un mayor riesgo de SRI en pacientes que inician tratamiento antirretroviral para el VIH incluyen SK más extenso (T1), una mayor carga viral del VIH, y el uso de TAR sin quimioterapia [47]. La progresión de SK en pacientes con SRI puede ser grave y se ha asociado con la muerte en algunos casos [47-49].
TRATAMIENTO SINTOMÁTICO LOCAL
Las modalidades de tratamiento local son útiles para la cosmética o la gestión de las lesiones de SK voluminoso sintomáticos, pero que no impiden el desarrollo de nuevas lesiones en áreas no tratadas.
Los enfoques de tratamiento locales más utilizados son:
1 Quimioterapia intralesional: la quimioterapia intralesional puede inducir la regresión de los tumores inyectados y se prefiere para lesiones pequeñas.
La vinblastina es el agente más ampliamente utilizado. Se puede inyectar directamente en la lesión SK como una solución de 0,2 a 0,3 mg / ml con un volumen de 0,1 ml por 0,5 cm2 de la lesión. Múltiples inyecciones pueden ser necesarias para lesiones más grandes. Una segunda serie de inyecciones a menudo es necesario tres a cuatro semanas más tarde. Lesiones tratadas se desvanecen y retroceder aunque normalmente se resuelve por completo [50-52]. En una serie de 42 pacientes con SK oral, por ejemplo, 74 por ciento mostró una reducción de más del 50 por ciento en las lesiones; La paliación se logró durante una media de 4,3 meses [50].
2 Radioterapia: La función principal de la radioterapia es el tratamiento de la enfermedad sintomática que es demasiado extensa para ser tratada con quimioterapia intralesional [53], pero no es lo suficientemente importante como para requerir tratamiento sistémico. Aunque las molestias de la radioterapia son frecuentes, por lo general se resuelve dentro de dos semanas de tratamiento. La radioterapia no tiene un papel en pacientes con lesiones de SK extensas, como se ilustra en un ensayo aleatorio de Zimbabwe [54]. En este ensayo, la radioterapia no mejoró la calidad de vida o la supervivencia en comparación con la atención de apoyo general sola en 495 pacientes que no fueron tratados con medicamentos antirretrovirales.
Alitretinoína tópica: La alitretinoína (ácido 9-cis retinoico) está disponible como un gel tópico que el paciente puede aplicarse para tratar las lesiones cutáneas del SK. Este agente se utiliza muy poco, ya que el gel tópico puede causar inflamación y conducir a cambios en la pigmentación en los pacientes de piel oscura
Quimioterapia sistémica: quimioterapia sistémica se utiliza generalmente para los pacientes con SK más avanzados o cuando hay evidencia de progresión rápida de la enfermedad [34]. Cuando está indicada la quimioterapia, el tratamiento con doxorubicina liposomal pegilada o daunorrubicina liposomal en general se recomienda como tratamiento de primera línea para SK [34]. Otros agentes que se han utilizado incluyen paclitaxel, bleomicina, vinblastina, vincristina y etopósido [57].
Generalmente se acepta que las indicaciones para añadir quimioterapia sistémica a la TAR incluyen:
- Afectación cutánea generalizada (por ejemplo, más de 25 lesiones)
- SK cutáneo extenso que no responde al tratamiento local
- Linfedemadema extenso
- Afectación visceral sintomática
- Síndrome de reconstitución inmune (SRI)
Antraciclinas liposomales: El tratamiento con doxorubicina liposomal pegilada o daunorrubicina liposomal se recomiendan generalmente como el tratamiento de primera línea para el SK relacionado con el SIDA cuando se indica la quimioterapia sistémica [34].
Las antraciclinas pegiladas tienen una vida media plasmática más larga que las formulaciones no liposomales. Las formulaciones liposomales tienen menos toxicidad en órganos no diana que las antraciclinas convencionales y proporcionan la ventaja teórica de concentraciones más altas de fármaco tumorales. Pueden reducir el tamaño los tumores, disminuir el edema, y hacer que el color de las lesiones desaparezca. Las tasas de respuesta varían del 30 al 60 por ciento dependiendo de la definición de respuesta clínica y las características específicas de los diferentes ensayos.
Los efectos secundarios de estos productos liposomales son en general leves. La alopecia y neuropatías son inusuales con estas preparaciones, en contraste con el perfil de efectos secundarios de los regímenes de quimioterapia de combinación convencionales. La cardiotoxicidad disminuida con antraciclinas liposomales permite mayores dosis acumuladas de antraciclina a administrar, el alargamiento de la duración en el que se pueden utilizar estos agentes [62,64].
El potencial para el control prolongado de SK fue ilustrado por un estudio de 98 pacientes tratados con TAR más doxorrubicina liposomal pegilada entre 1997 y 2002 [65]. Con una mediana de seguimiento de 29 meses, 29 pacientes (30 por ciento) habían muerto, incluyendo tres con SK progresivos. Entre los 61 pacientes que tuvieron una respuesta completa o parcial de sus SK, sólo ocho (13 por ciento) tuvieron recaídas después de terminar la quimioterapia. La duración óptima del tratamiento con antraciclina liposomal es incierto. Un curso de cuatro a seis ciclos de doxorrubicina liposomal seguido por un periodo de observación puede ser razonable.
Taxanos : Aunque paclitaxel es potencialmente más tóxico que las antraciclinas liposomales, tiene gran eficacia como tratamiento de segunda línea para SK [66-70], y pueden ser una alternativa para el tratamiento inicial de los pacientes con SK sintomático avanzado.
La administración de paclitaxel requiere premedicación con glucocorticoides. Debido a la preocupación de que los esteroides pueden promover inmunosuprimir pacientes infectados por el VIH y exacerbar el SK, la dosis de dexametasona se reduce típicamente a 10 mg por vía oral 12 y 6 horas antes de la quimioterapia, en lugar de la dosis habitual de 20 mg. Paclitaxel se metaboliza a través de las enzimas del citocromo P450, al igual que muchos de los medicamentos antirretrovirales
Esteroides: El tratamiento con corticoides se ha asociado con la inducción de SK y con la exacerbación de SK preexistentes en personas infectadas por el VIH, así como en pacientes sin SIDA que reciben corticosteroides para trasplante de órganos, enfermedades autoinmunes o enfermedades linfoproliferativos [74]. La asociación de corticosteroides con SK es importante debido a la frecuente utilización de estos agentes en pacientes infectados por el VIH con una variedad de trastornos, incluyendo la trombocitopenia inmune (PTI) y la neumonía por Pneumocystis jirovecii.
En los pacientes en tratamiento con esteroides, las lesiones de SK pueden retroceder tras la reducción o retirada de esteroides [74,75].
Otros agentes: Un número de otros agentes se han estudiado de forma más limitada en pacientes con sarcoma de Kaposi relacionado con el SIDA, y éstos pueden tener utilidad como terapia de segunda línea:
- Vinorelbina - vinorelbina puede ser eficaz en el tratamiento de pacientes con SK relacionado al SIDA que han fracasado otros tratamientos, incluyendo otros alcaloides de la vinca. En una serie de 35 pacientes tratados con vinorelbina (30 mg / m 2 cada dos semanas), tres (9 por ciento) tuvieron una respuesta clínica completa, y 12 (34 por ciento) tuvieron una respuesta parcial, que se prolongó durante una media de 151 días [76].
- Etopósido: Etopósido oral se ha informado que tiene actividad en el SK relacionado con el SIDA cuando se les da ya sea en dosis fraccionadas (20 mg / m 2 cada ocho horas durante 7 de 21 días) [77] o como una dosis una vez al día de 50 mg por 7 de cada 21 días [78].
- Interferón-alfa - interferón-alfa (IFNa) es un modificador de la respuesta biológica que produce respuestas clínicamente significativas en aproximadamente 20 a 40 por ciento de los pacientes con sarcoma de Kaposi relacionado con el SIDA, especialmente aquellos con enfermedad limitada a la piel e inmunosupresión relativamente modesta [79.80]. Sin embargo, IFNa se utiliza muy poco debido a la disponibilidad de otros agentes con una relación beneficio-riesgo más favorable.
- Imatinib: La activación del factor de crecimiento derivado de plaquetas (PDGF) y los receptores de c-kit (ambos receptores tirosina quinasas) son importantes en el crecimiento de las lesiones de SK, e imatinib inhibe estos receptores. En un estudio multicéntrico de fase II, 30 pacientes fueron tratados con imatinib durante un máximo de 12 meses [83].
- Bevacizumab: Bevacizumab es un anticuerpo monoclonal dirigido contra el factor de crecimiento endotelial vascular, lo que contribuye a la patogénesis de SK. En un estudio de fase II, el tratamiento de 17 pacientes con VIH asociada SK usando bevacizumab resultó en una respuesta objetiva (completa o parcial) en cinco casos y la enfermedad estable en nueve casos adicionales [86].
Estudios de casos y controles de cohortes históricas de sujetos seropositivos al VIH sugieren que hay una menor incidencia de SK en los pacientes tratados con ganciclovir o foscarnet, pero no aciclovir [96,97
PRONÓSTICO
La combinación de TAR y quimioterapia ha dado lugar a una notable disminución en la incidencia de SK y una mejora en el pronóstico de los pacientes con SK relacionado con el VIH, lo que con frecuencia se habían asociado con la muerte temprana.
Fuente: UpToDate 2014
Bibliografía
1 Moore PS, Chang Y. Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. N Engl J Med 1995; 332:1181.
2 Beral V, Peterman TA, Berkelman RL, Jaffe HW. Kaposi's sarcoma among persons with AIDS: a sexually transmitted infection? Lancet 1990; 335:123.
3 Stefan DC, Stones DK, Wainwright L, Newton R. Kaposi sarcoma in South African children. Pediatr Blood Cancer 2011; 56:392.
4 Senba M, Buziba N, Mori N, et al. Increased prevalence of Kaposi΄s sarcoma-associated herpesvirus in the Kaposi΄s sarcoma-endemic area of western Kenya in 1981-2000. Acta Virol 2011; 55:161.
5 Iscovich J, Boffetta P, Winkelmann R, et al. Classic Kaposi's sarcoma in Jews living in Israel, 1961-1989: a population-based incidence study. AIDS 1998; 12:2067.
6 Fenig E, Brenner B, Rakowsky E, et al. Classic Kaposi sarcoma: experience at Rabin Medical Center in Israel. Am J Clin Oncol 1998; 21:498.
7 Lodi S, Guiguet M, Costagliola D, et al. Kaposi sarcoma incidence and survival among HIV-infected homosexual men after HIV seroconversion. J Natl Cancer Inst 2010; 102:784.
8 Ledergerber B, Telenti A, Egger M. Risk of HIV related Kaposi's sarcoma and non-Hodgkin's lymphoma with potent antiretroviral therapy: prospective cohort study. Swiss HIV Cohort Study. BMJ 1999; 319:23.
9 International Collaboration on HIV and Cancer. Highly active antiretroviral therapy and incidence of cancer in human immunodeficiency virus-infected adults. J Natl Cancer Inst 2000; 92:1823.
10 Mocroft A, Kirk O, Clumeck N, et al. The changing pattern of Kaposi sarcoma in patients with HIV, 1994-2003: the EuroSIDA Study. Cancer 2004; 100:2644.
11 Engels EA, Pfeiffer RM, Goedert JJ, et al. Trends in cancer risk among people with AIDS in the United States 1980-2002. AIDS 2006; 20:1645.
12 Eltom MA, Jemal A, Mbulaiteye SM, et al. Trends in Kaposi's sarcoma and non-Hodgkin's lymphoma incidence in the United States from 1973 through 1998. J Natl Cancer Inst 2002; 94:1204.
13 Yanik EL, Napravnik S, Cole SR, et al. Incidence and timing of cancer in HIV-infected individuals following initiation of combination antiretroviral therapy. Clin Infect Dis 2013; 57:756.
14 Osmond DH, Buchbinder S, Cheng A, et al. Prevalence of Kaposi sarcoma-associated herpesvirus infection in homosexual men at beginning of and during the HIV epidemic. JAMA 2002; 287:221.
15 Martin JN, Ganem DE, Osmond DH, et al. Sexual transmission and the natural history of human herpesvirus 8 infection. N Engl J Med 1998; 338:948.
16 Rezza G, Andreoni M, Dorrucci M, et al. Human herpesvirus 8 seropositivity and risk of Kaposi's sarcoma and other acquired immunodeficiency syndrome-related diseases. J Natl Cancer Inst 1999; 91:1468.
17 Sitas F, Carrara H, Beral V, et al. Antibodies against human herpesvirus 8 in black South African patients with cancer. N Engl J Med 1999; 340:1863.
18 Trattner A, Hodak E, David M, Sandbank M. The appearance of Kaposi sarcoma during corticosteroid therapy. Cancer 1993; 72:1779.
19 Gill PS, Loureiro C, Bernstein-Singer M, et al. Clinical effect of glucocorticoids on Kaposi sarcoma related to the acquired immunodeficiency syndrome (AIDS). Ann Intern Med 1989; 110:937.
20 Litwin MA, Williams CM. Cutaneous Pneumocystis carinii infection mimicking Kaposi sarcoma. Ann Intern Med 1992; 117:48.
21 Mitsuyasu RT. Clinical variants and staging of Kaposi's sarcoma. Semin Oncol 1987; 14:13.
22 Ioachim HL, Adsay V, Giancotti FR, et al. Kaposi's sarcoma of internal organs. A multiparameter study of 86 cases. Cancer 1995; 75:1376.
23 Caponetti G, Dezube BJ, Restrepo CS, Pantanowitz L. Kaposi sarcoma of the musculoskeletal system: a review of 66 patients. Cancer 2007; 109:1040.
24 Bower M, Dalla Pria A, Coyle C, et al. Prospective stage-stratified approach to AIDS-related Kaposi's sarcoma. J Clin Oncol 2014; 32:409.
25 Nichols CM, Flaitz CM, Hicks MJ. Treating Kaposi's lesions in the HIV-infected patient. J Am Dent Assoc 1993; 124:78.
26 Danzig JB, Brandt LJ, Reinus JF, Klein RS. Gastrointestinal malignancy in patients with AIDS. Am J Gastroenterol 1991; 86:715.
27 Laine L, Amerian J, Rarick M, et al. The response of symptomatic gastrointestinal Kaposi's sarcoma to chemotherapy: a prospective evaluation using an endoscopic method of disease quantification. Am J Gastroenterol 1990; 85:959.
28 Friedman SL, Wright TL, Altman DF. Gastrointestinal Kaposi's sarcoma in patients with acquired immunodeficiency syndrome. Endoscopic and autopsy findings. Gastroenterology 1985; 89:102.
29 Dezube BJ. Clinical presentation and natural history of AIDS--related Kaposi's sarcoma. Hematol Oncol Clin North Am 1996; 10:1023.
30 Holland JC, Tross S. Psychosocial considerations in the therapy of epidemic Kaposi's sarcoma. Semin Oncol 1987; 14:48.
31 Krown SE, Metroka C, Wernz JC. Kaposi's sarcoma in the acquired immune deficiency syndrome: a proposal for uniform evaluation, response, and staging criteria. AIDS Clinical Trials Group Oncology Committee. J Clin Oncol 1989; 7:1201.
32 El Amari EB, Toutous-Trellu L, Gayet-Ageron A, et al. Predicting the evolution of Kaposi sarcoma, in the highly active antiretroviral therapy era. AIDS 2008; 22:1019.
33 Dezube BJ, Pantanowitz L, Aboulafia DM. Management of AIDS-related Kaposi sarcoma: advances in target discovery and treatment. AIDS Read 2004; 14:236.
34 Bower M, Collins S, Cottrill C, et al. British HIV Association guidelines for HIV-associated malignancies 2008. HIV Med 2008; 9:336.
35 Krown SE. Highly active antiretroviral therapy in AIDS-associated Kaposi's sarcoma: implications for the design of therapeutic trials in patients with advanced, symptomatic Kaposi's sarcoma. J Clin Oncol 2004; 22:399.
36 Stebbing J, Sanitt A, Nelson M, et al. A prognostic index for AIDS-associated Kaposi's sarcoma in the era of highly active antiretroviral therapy. Lancet 2006; 367:1495.
37 Grabar S, Abraham B, Mahamat A, et al. Differential impact of combination antiretroviral therapy in preventing Kaposi's sarcoma with and without visceral involvement. J Clin Oncol 2006; 24:3408.
38 Franceschi S, Maso LD, Rickenbach M, et al. Kaposi sarcoma incidence in the Swiss HIV Cohort Study before and after highly active antiretroviral therapy. Br J Cancer 2008; 99:800.
39 Mocroft A, Kirk O, Clumeck N, et al. The changing pattern of Kaposi sarcoma in patients with HIV, 1994-2003: the EuroSIDA Study. Cancer 2004; 100:2644.
40 Gill J, Bourboulia D, Wilkinson J, et al. Prospective study of the effects of antiretroviral therapy on Kaposi sarcoma--associated herpesvirus infection in patients with and without Kaposi sarcoma. J Acquir Immune Defic Syndr 2002; 31:384.
41 Gallafent JH, Buskin SE, De Turk PB, Aboulafia DM. Profile of patients with Kaposi's sarcoma in the era of highly active antiretroviral therapy. J Clin Oncol 2005; 23:1253.
42 Sgadari C, Barillari G, Toschi E, et al. HIV protease inhibitors are potent anti-angiogenic molecules and promote regression of Kaposi sarcoma. Nat Med 2002; 8:225.
43 Martinez V, Caumes E, Gambotti L, et al. Remission from Kaposi's sarcoma on HAART is associated with suppression of HIV replication and is independent of protease inhibitor therapy. Br J Cancer 2006; 94:1000.
44 Krown SE, Lee JY, Dittmer DP, AIDS Malignancy Consortium. More on HIV-associated Kaposi's sarcoma. N Engl J Med 2008; 358:535.
45 Bower M, Nelson M, Young AM, et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J Clin Oncol 2005; 23:5224.
46 Leidner RS, Aboulafia DM. Recrudescent Kaposi's sarcoma after initiation of HAART: a manifestation of immune reconstitution syndrome. AIDS Patient Care STDS 2005; 19:635.
47 Letang E, Lewis JJ, Bower M, et al. Immune reconstitution inflammatory syndrome associated with Kaposi sarcoma: higher incidence and mortality in Africa than in the UK. AIDS 2013; 27:1603.
48 Achenbach CJ, Harrington RD, Dhanireddy S, et al. Paradoxical immune reconstitution inflammatory syndrome in HIV-infected patients treated with combination antiretroviral therapy after AIDS-defining opportunistic infection. Clin Infect Dis 2012; 54:424.
49 Stover KR, Molitorisz S, Swiatlo E, Muzny CA. A fatal case of kaposi sarcoma due to immune reconstitution inflammatory syndrome. Am J Med Sci 2012; 343:421.
50 Epstein JB. Treatment of oral Kaposi sarcoma with intralesional vinblastine. Cancer 1993; 71:1722.
51 McCormick SU. Intralesional vinblastine injections for the treatment of oral Kaposi's sarcoma: report of 10 patients with 2-year follow-up. J Oral Maxillofac Surg 1996; 54:583.
52 Ramírez-Amador V, Esquivel-Pedraza L, Lozada-Nur F, et al. Intralesional vinblastine vs. 3% sodium tetradecyl sulfate for the treatment of oral Kaposi's sarcoma. A double blind, randomized clinical trial. Oral Oncol 2002; 38:460.
53 Donato V, Guarnaccia R, Dognini J, et al. Radiation therapy in the treatment of HIV-related Kaposi's sarcoma. Anticancer Res 2013; 33:2153.
54 Olweny CL, Borok M, Gudza I, et al. Treatment of AIDS-associated Kaposi's sarcoma in Zimbabwe: results of a randomized quality of life focused clinical trial. Int J Cancer 2005; 113:632.
55 Walmsley S, Northfelt DW, Melosky B, et al. Treatment of AIDS-related cutaneous Kaposi's sarcoma with topical alitretinoin (9-cis-retinoic acid) gel. Panretin Gel North American Study Group. J Acquir Immune Defic Syndr 1999; 22:235.
56 Bodsworth NJ, Bloch M, Bower M, et al. Phase III vehicle-controlled, multi-centered study of topical alitretinoin gel 0.1% in cutaneous AIDS-related Kaposi's sarcoma. Am J Clin Dermatol 2001; 2:77.
57 Lee FC, Mitsuyasu RT. Chemotherapy of AIDS--related Kaposi's sarcoma. Hematol Oncol Clin North Am 1996; 10:1051.
58 Mosam A, Shaik F, Uldrick TS, et al. A randomized controlled trial of highly active antiretroviral therapy versus highly active antiretroviral therapy and chemotherapy in therapy-naive patients with HIV-associated Kaposi sarcoma in South Africa. J Acquir Immune Defic Syndr 2012; 60:150.
59 Martin-Carbonero L, Barrios A, Saballs P, et al. Pegylated liposomal doxorubicin plus highly active antiretroviral therapy versus highly active antiretroviral therapy alone in HIV patients with Kaposi's sarcoma. AIDS 2004; 18:1737.
60 Northfelt DW, Dezube BJ, Thommes JA, et al. Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi's sarcoma: results of a randomized phase III clinical trial. J Clin Oncol 1998; 16:2445.
61 Stewart S, Jablonowski H, Goebel FD, et al. Randomized comparative trial of pegylated liposomal doxorubicin versus bleomycin and vincristine in the treatment of AIDS-related Kaposi's sarcoma. International Pegylated Liposomal Doxorubicin Study Group. J Clin Oncol 1998; 16:683.
62 Gill PS, Wernz J, Scadden DT, et al. Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi's sarcoma. J Clin Oncol 1996; 14:2353.
63 Cooley T, Henry D, Tonda M, et al. A randomized, double-blind study of pegylated liposomal doxorubicin for the treatment of AIDS-related Kaposi's sarcoma. Oncologist 2007; 12:114.
64 Young AM, Dhillon T, Bower M. Cardiotoxicity after liposomal anthracyclines. Lancet Oncol 2004; 5:654.
65 Martín-Carbonero L, Palacios R, Valencia E, et al. Long-term prognosis of HIV-infected patients with Kaposi sarcoma treated with pegylated liposomal doxorubicin. Clin Infect Dis 2008; 47:410.
66 Welles L, Saville MW, Lietzau J, et al. Phase II trial with dose titration of paclitaxel for the therapy of human immunodeficiency virus-associated Kaposi's sarcoma. J Clin Oncol 1998; 16:1112.
67 Gill PS, Tulpule A, Espina BM, et al. Paclitaxel is safe and effective in the treatment of advanced AIDS-related Kaposi's sarcoma. J Clin Oncol 1999; 17:1876.
68 Tulpule A, Groopman J, Saville MW, et al. Multicenter trial of low-dose paclitaxel in patients with advanced AIDS-related Kaposi sarcoma. Cancer 2002; 95:147.
69 Lee, FC, Thornton, K, Williams, B. Low dose weekly paclitaxel is an effective first line treatment for patients with symptomatic AIDS-KS (abstract). Proc Am Soc Clin Oncol 2003; 22:825a.
70 Lim ST, Tupule A, Espina BM, Levine AM. Weekly docetaxel is safe and effective in the treatment of advanced-stage acquired immunodeficiency syndrome-related Kaposi sarcoma. Cancer 2005; 103:417.
71 Cianfrocca M, Lee S, Von Roenn J, et al. Randomized trial of paclitaxel versus pegylated liposomal doxorubicin for advanced human immunodeficiency virus-associated Kaposi sarcoma: evidence of symptom palliation from chemotherapy. Cancer 2010; 116:3969.
72 Schwartz JD, Howard W, Scadden DT. Potential interaction of antiretroviral therapy with paclitaxel in patients with AIDS-related Kaposi's sarcoma. AIDS 1999; 13:283.
73 Nannan Panday VR, Hoetelmans RM, van Heeswijk RP, et al. Paclitaxel in the treatment of human immunodeficiency virus 1-associated Kaposi's sarcoma--drug-drug interactions with protease inhibitors and a nonnucleoside reverse transcriptase inhibitor: a case report study. Cancer Chemother Pharmacol 1999; 43:516.
74 Trattner A, Hodak E, David M, Sandbank M. The appearance of Kaposi sarcoma during corticosteroid therapy. Cancer 1993; 72:1779.
75 Gill PS, Loureiro C, Bernstein-Singer M, et al. Clinical effect of glucocorticoids on Kaposi sarcoma related to the acquired immunodeficiency syndrome (AIDS). Ann Intern Med 1989; 110:937.
76 Nasti G, Errante D, Talamini R, et al. Vinorelbine is an effective and safe drug for AIDS-related Kaposi's sarcoma: results of a phase II study. J Clin Oncol 2000; 18:1550.
77 Sprinz E, Caldas AP, Mans DR, et al. Fractionated doses of oral etoposide in the treatment of patients with aids-related kaposi sarcoma: a clinical and pharmacologic study to improve therapeutic index. Am J Clin Oncol 2001; 24:177.
78 Evans SR, Krown SE, Testa MA, et al. Phase II evaluation of low-dose oral etoposide for the treatment of relapsed or progressive AIDS-related Kaposi's sarcoma: an AIDS Clinical Trials Group clinical study. J Clin Oncol 2002; 20:3236.
79 Krown SE, Li P, Von Roenn JH, et al. Efficacy of low-dose interferon with antiretroviral therapy in Kaposi's sarcoma: a randomized phase II AIDS clinical trials group study. J Interferon Cytokine Res 2002; 22:295.
80 Shepherd FA, Beaulieu R, Gelmon K, et al. Prospective randomized trial of two dose levels of interferon alfa with zidovudine for the treatment of Kaposi's sarcoma associated with human immunodeficiency virus infection: a Canadian HIV Clinical Trials Network study. J Clin Oncol 1998; 16:1736.
81 Karp JE, Pluda JM, Yarchoan R. AIDS-related Kaposi's sarcoma. A template for the translation of molecular pathogenesis into targeted therapeutic approaches. Hematol Oncol Clin North Am 1996; 10:1031.
82 McGarvey ME, Tulpule A, Cai J, et al. Emerging treatments for epidemic (AIDS-related) Kaposi's sarcoma. Curr Opin Oncol 1998; 10:413.
83 Koon HB, Krown SE, Lee JY, et al. Phase II Trial of Imatinib in AIDS-Associated Kaposi’s Sarcoma: AIDS Malignancy Consortium Protocol 042. J Clin Oncol.
84 Stallone G, Schena A, Infante B, et al. Sirolimus for Kaposi's sarcoma in renal-transplant recipients. N Engl J Med 2005; 352:1317.
85 Krown SE, Roy D, Lee JY, et al. Rapamycin with antiretroviral therapy in AIDS-associated Kaposi sarcoma: an AIDS Malignancy Consortium study. J Acquir Immune Defic Syndr 2012; 59:447.
86 Uldrick TS, Wyvill KM, Kumar P, et al. Phase II study of bevacizumab in patients with HIV-associated Kaposi's sarcoma receiving antiretroviral therapy. J Clin Oncol 2012; 30:1476.
87 Masood R, Nagpal S, Zheng T, et al. Kaposi sarcoma is a therapeutic target for vitamin D(3) receptor agonist. Blood 2000; 96:3188.
88 Dezube BJ, Von Roenn JH, Holden-Wiltse J, et al. Fumagillin analog in the treatment of Kaposi's sarcoma: a phase I AIDS Clinical Trial Group study. AIDS Clinical Trial Group No. 215 Team. J Clin Oncol 1998; 16:1444.
89 Little RF, Wyvill KM, Pluda JM, et al. Activity of thalidomide in AIDS-related Kaposi's sarcoma. J Clin Oncol 2000; 18:2593.
90 Miles SA, Dezube BJ, Lee JY, et al. Antitumor activity of oral 9-cis-retinoic acid in HIV-associated Kaposi's sarcoma. AIDS 2002; 16:421.
91Dezube BJ, Krown SE, Lee JY, et al. Randomized phase II trial of matrix metalloproteinase inhibitor COL-3 in AIDS-related Kaposi's sarcoma: an AIDS Malignancy Consortium Study. J Clin Oncol 2006; 24:1389.
92 Little RF, Pluda JM, Wyvill KM, et al. Activity of subcutaneous interleukin-12 in AIDS-related Kaposi sarcoma. Blood 2006; 107:4650.
93 Little RF, Aleman K, Kumar P, et al. Phase 2 study of pegylated liposomal doxorubicin in combination with interleukin-12 for AIDS-related Kaposi sarcoma. Blood 2007; 110:4165.
94 Gill PS, Lunardi-Ishkandar Y, Louie S, et al. The effects of preparations of human chorionic gonadotropin on AIDS-related Kaposi's sarcoma. N Engl J Med 1996; 335:1261.
95 Gill PS, McLaughlin T, Espina BM, et al. Phase I study of human chorionic gonadotropin given subcutaneously to patients with acquired immunodeficiency syndrome-related mucocutaneous Kaposi's sarcoma. J Natl Cancer Inst 1997; 89:1797.
96 Mocroft A, Youle M, Gazzard B, et al. Anti-herpesvirus treatment and risk of Kaposi's sarcoma in HIV infection. Royal Free/Chelsea and Westminster Hospitals Collaborative Group. AIDS 1996; 10:1101.
97 Glesby MJ, Hoover DR, Weng S, et al. Use of antiherpes drugs and the risk of Kaposi's sarcoma: data from the Multicenter AIDS Cohort Study. J Infect Dis 1996; 173:1477.
98 Martin DF, Kuppermann BD, Wolitz RA, et al. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N Engl J Med 1999; 340:1063.
99 Bower M, Dalla Pria A, Coyle C, et al. Prospective stage-stratified approach to AIDS-related Kaposi's sarcoma. J Clin Oncol 2014; 32:409.